Die passende Antwort auf häufig gestellte Fragen
Finden Sie erklärende Informationen zu unseren Produkten und Dienstleistungen, indem Sie auf die folgenden Links klicken.
Next Generation Sequencing
Gibt es Besonderheiten bezüglich der Norm DIN EN ISO 17025?
Unter den unten aufgeführten Bedingungenkönnen auch nur verkürzte Prüfberichte erstellt werden.
Betrifft: Vereinfachte Prüfberichte nach DIN EN ISO 170252018, (Abschnitt 7.8)
Prüflabore (Akkreditierung): Eurofins Genomics Europe Applied Genomics GmbH (D-PL-13372, ) Eurofins Genomics Europe Sequencing GmbH (D-PL-17038)
Für akkreditierte Prüfungen bei diesen beiden Gesellschaften werden Prüfergebnisse ggf. nur vereinfacht berichtet, d.h. Anforderungen der Norm DIN EN ISO 17025:2018 (Abschnitt 7.8) sind für diese Art der Ergebnisübermittlung nicht vollumfänglich zutreffend.
Bei diesen Prüfungen können die Ergebnisse daher auch als Kurzprüfberichte, elektronisch erzeugten Result reports, Exceltabellen oder nur als Ergebnisdateien berichtet werden.
Bei Fragen dazu wenden Sie sich bitte an das Customer Care Team.
Fragen zu INVIEW Resequencing
Wo bekomme ich meine Ergebnisse?
Alle Rohdaten (FAST-Q Dateien) sowie die analysierten und assemblierten Daten können über Ihr sicheres Online-Konto heruntergeladen werden. Beispieldaten der De novo-Assemblierung von E. coli DH1 können in der Sektion "Dateien" eingesehen werden.
Wie soll ich meine Proben verschicken?
Bitte senden Sie aufgereinigte DNA/RNA bzw. Amplikons in 1,5 ml SnapCap Tubes (ohne Parafilm). Und kleben Sie den kostenfreien NGS Barcode quer auf das Tube.
Ab 92 Proben können Sie diese auch gerne in Platten schicken. Bitte verwenden Sie hierfür "Full-Skirted" PCR Platten (zBsp von Eppendorf oder Twin).
Für mehr Informationen und den Probenversand für Extraktionsproben lesen Sie auch unseren Sample Submission Guide.
(Im Bild werden Prepaid Sanger Barcodes verwendet, aber der Ablauf ist für die kostenfreien NGS Barcodes (blau) derselbe)
|
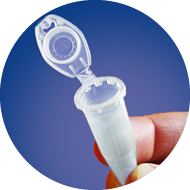
1
Only use 1.5 ml
safe-lock tubes
|

2
Remove barcode
stricker from film
|
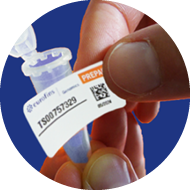
3
Affix barcode sticker
horizontally
|
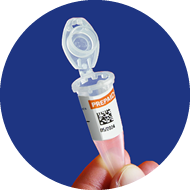
4
QR-code visible at front so
it can be read by scaner
|
Wohin soll ich meine Proben schicken?
Eurofins Genomics Europe Pharma and Diagnostics Products & Services Sanger/PCR GmbH
Jakob-Stadler-Platz 7
78467 Konstanz
Germany
Fragen zu INVIEW Whole Exome
Wie sieht der Prozess der Datenverarbeitung aus (einschließlich QCI Interpret Translational™ von QIAGEN)?
Alle Rohdaten werden analysiert und in eine VCF-Datei konvertiert, die direkt auf die webbasierte Software-Plattform von QIAGEN hochgeladen wird. Die Zugangsdaten zu Ihrem Konto erhalten Sie von Eurofins Genomics.
Darüber hinaus erfolgt die Lieferung der Alignment-Datei (BAM), der SNP- und InDel-Tabellen (VCF und TSV) und des Datenanalyseberichts (PDF) von Eurofins Genomics.
Bitte beachten: Der Kunde muss dem Endbenutzer-Lizenzvertrag von QIAGEN zustimmen, damit der Zugriff auf das Produkt Variant Analysis oder die von diesem Produkt generierten Ergebnisse gewährt wird.
Wie lange habe ich Zugriff auf meine Daten auf meinem QIAGEN-Konto?
Die Lizenz gewährt Ihnen einen sechsmonatigen Erstzugriff auf die Analysesoftware. Nach Ablauf dieses Zeitraums haben Sie die Möglichkeit, die Lizenz zu erneuern; die Erneuerung kann jedoch nur offline erworben werden. Bitte sprechen Sie uns an!
Wo bekomme ich meine Ergebnisse, wenn ich nur die optionale BioIT wähle?
Alle Rohdaten (FAST-Q Dateien) sowie die analysierten Daten können über Ihr gesichertes Konto heruntergeladen werden.
Was für eine Abdeckung soll ich verwenden?
Zum Auffinden von SNPs in heterozygoten Organismen empfehlen wir mindestens eine 30x-Abdeckung. Eine höhere Abdeckung erhöht die Zuverlässigkeit der SNP-Auffindung. Eine zuverlässige Aufdeckung von genetischen Varianten in inhomogenen Proben wie etwa aus normalen Zellen und Tumorzellen extrahierter DNA erfordert eine höhere Gesamtabdeckung. Die zum Erreichen einer bestimmten Abdeckung auf der interessierenden DNA benötigte Datenmenge ist vom Verhältnis von normaler DNA zu Tumor-DNA abhängig. Bitte beachten Sie, dass auf Grund der unterschiedlichen Effizienz der für die Anreicherung verwendeten Baits die Zielregionen nicht gleichmäßig abgedeckt werden (siehe unten).
Wie handhaben Sie PCR-Duplikate?
Die Häufigkeit von PCR-Duplikaten ist direkt mit Qualität und Menge des Ausgangsmaterials korreliert. Die Library-Erstellung mit weniger als der empfohlenen Menge an DNA erfordert zusätzliche PCR-Zyklen, damit genügend Material zum Beladen des Sequenziergeräts erzeugt wird. Die Häufigkeit von PCR-Duplikaten hängt daher von der Probe ab und kann von GATC Biotech nicht beeinflusst werden.
Duplikate sind aus der Berechnung der durchschnittlichen zielgerichteten Abdeckung nicht ausgeschlossen. Gemäß unserer Erfahrung mit der Sequenzierung humaner Proben erzielen wir für qualitativ hochwertige DNA in der Regel PCR-Duplikatraten von ca. 5 %.
Wenn weitere bioinformatische Analysen (z. B. SNP-Identifizierung) in Auftrag gegeben werden, dann wird nur eine Kopie des doppelten Read-Paares im Alignment behalten. Der Rest wird zur Vermeidung eines Bias aus der weiteren Analyse ausgenommen.
Was für ein Ausgangsmaterial soll ich schicken? In welchen Mengen?
Für optimale Ergebnisse benötigen wir mindestens 100 ng aufgereinigte RNA-freie Doppelstrang-DNA mit hohem Molekulargewicht (Konzentration ≥ 1 ng/µl; OD 260/280 ≥ 1,8; OD 260/230 ≥ 2,0). Für DNA von FFPE-Proben benötigen wir mindestens 200 ng DNA.
Eine Kontamination durch DNA von anderen Spezies (> 3 %, insbesondere von nah verwandten Spezies) könnte die Anreicherung der humanen DNA stören und reduziert zudem die Gesamtmenge an humaner DNA in der gesendeten Probe. Akzeptiert wird in der Regel amplifizierte Gesamtgenom (WGA)-DNA oder DNA aus archiviertem Gewebe, wie beispielsweise Formalin-fixierte, Paraffin-eingebettete (FFPE) Proben. Wenden Sie sich an uns, wenn Sie Einzelheiten wissen möchten.
Was meinen Sie mit überlappenden Reads?
Die Fragmentierung genomischer DNA nach dem Agilent SureSelectXT-Protokoll und die darauffolgende Weiterverarbeitung ergeben eine Normalverteilung von DNA-Fragmenten mit unterschiedlichen Längen. Ein kleiner Prozentsatz der entstandenen Library-Fragmente haben eine Insert-Größe von weniger als 250 bp. Die Sequenzierung dieser Library-Fragmente mit Paired-End-Reads von 150 bp erzeugt teilweise überlappende Reads, welche die gleichen Basen abdecken; somit entsteht eine künstliche Verdoppelung der Abdeckung an diesen Stellen. Zur Gewährleistung präziser Analysen werden diese Basen aus weiteren Analysen (z. B. Erkennung von Einzelnukleotid-Varianten und Insertionen/Deletionen) ausgeschlossen. Darüber hinaus werden die Protokolle bei Eurofins Genomics fortlaufend mit dem Ziel optimiert, die Anzahl an überlappenden Basen auf ein Minimum zu reduzieren.
Wie sieht die Qualität der Daten aus?
Bei der Sequenzierung von humanen Proben im Paired-End-Modus (150 bp) erzielt Eurofins Genomics in der Regel über 80 % Basecalls mit einem Qualitätswert von mehr als Q30 (> 99,9 % Genauigkeit).
Garantieren Sie einen bestimmten zielgenauen Output?
Für Proben, die die erste Qualitätskontrolle bei Eurofins Genomics erfolgreich bestanden haben, garantieren wir Ihnen den Ziel-Output, den Sie bestellt haben. Die durchschnittliche zielgenaue Abdeckung wird berechnet als Summe der kartierten Basen in jeder Zielposition dividiert durch die Anzahl der Basen in der Zielregion (Zielbasen / Größe der Zielregion).
Welche Gene werden angereichert?
Die Zielregion entspricht den Bait-Koordinaten des SureSelectHuman All Exon V6 Kits von Agilent: ca. 60 Mbp der exonischen Basen (> 93 % der Ziele werden mit > 20x, 90 % der Gene werden vollständig abgedeckt – alle Exons – 10 % der Gene werden teilweise abgedeckt).
Werden alle Gene bei 30x abgedeckt?
Auf Grund der unterschiedlichen Effizienz der zur Anreicherung verwendeten Baits (z. B. GC/AT-Gehalt) werden Zielregionen unterschiedlich abgedeckt und die Reichweite und Einheitlichkeit der Abdeckung schwanken innerhalb der Zielregion. Eurofins Genomics wendet das neueste Human All Exon Kit-Design (V6) von Agilent an, das verbesserte Design-Algorithmen aufweist und somit schwierige Regionen besser erfassen und eine hervorragende Einheitlichkeit der Abdeckung bieten kann.
Welche Art von Qualitätskontrollen führen Sie durch?
Qualität und Quantität jeder eingehenden Probe werden mit entsprechenden Methoden bestimmt. Weitere Qualitätskontrollen erfolgen bei verschiedenen Schritten des Prozesses.
Wohin soll ich meine Proben schicken?
Eurofins Genomics Europe Pharma and Diagnostics Products & Services Sanger/PCR GmbH
Jakob-Stadler-Platz 7
78467 Konstanz
Germany
Wie soll ich meine Proben verschicken?
Bitte senden Sie aufgereinigte DNA/RNA bzw. Amplikons in 1,5 ml SnapCap Tubes (ohne Parafilm). Und kleben Sie den kostenfreien NGS Barcode quer auf das Tube.
Ab 92 Proben können Sie diese auch gerne in Platten schicken. Bitte verwenden Sie hierfür "Full-Skirted" PCR Platten (zBsp von Eppendorf oder Twin).
Für mehr Informationen und den Probenversand für Extraktionsproben lesen Sie auch unseren Sample Submission Guide.
(Im Bild werden Prepaid Sanger Barcodes verwendet, aber der Ablauf ist für die kostenfreien NGS Barcodes (blau) derselbe)
|
1
Only use 1.5 ml
safe-lock tubes
|
2
Remove barcode
stricker from film
|
3
Affix barcode sticker
horizontally
|
4
QR-code visible at front so
it can be read by scaner
|
Fragen zu INVIEW Core Exome
Welche Art von Proben kann mit INVIEW Core Exome (37 Mb) verarbeitet werden?
Als Ausgangsmaterial für INVIEW Core Exome (37 Mb) akzeptieren wir Gewebe, Zellen und Blut. Amplifizierte Gesamtgenom (WGA)-DNA oder DNA aus archiviertem Gewebe, wie beispielsweise Formalin-fixierte, Paraffin-eingebettete (FFPE) Proben, werden in der Regel ebenfalls akzeptiert. Wenden Sie sich an uns, wenn Sie Einzelheiten wissen möchten.
Wie viel Ausgangsmaterial soll ich versenden?
Die Datengenerierung gilt nur für frisch isolierte, nicht amplifizierte und reine humane DNA-Proben (mindestens 200 ng). Bitte beachten Sie, dass Verunreinigungen durch DNA anderer Spezies (> 3 %; insbesondere von eng verwandten Organismen) die Gesamtmenge an humaner DNA in der gelieferten Probe reduziert und die Anreicherung der Exon-Region stören könnte.
Welche Gene werden angereichert?
Für die bestmögliche Abdeckung (> 99%) der proteinkodierenden Regionen des Humangenoms, wird eine Kombination aus 2 Panels verwendet. DAs Twist Human Core Exome (33 Mb) und das Twist Human RefSeq Panel (3.5 Mb)
Wie werden bei Ihnen PCR-Duplikate gehandhabt?
Die Häufigkeit von PCR-Duplikaten ist direkt mit Qualität und Menge des zur Verfügung gestellten Ausgangsmaterials korreliert.
Aufgrund unserem voll-automatisiertenBibliotheken-Herstellungsprozesses (Probentyp abhängiger DNA Input, Anpassung der PCR Zyklen) erhalten wir allerdings die geringstmögliche Anzahl an PCR-Duplikatraten. Die segmentierten "Flow cells" des HiSeq4000 und des NovaSeq6000, können allerdings zu auch zu PCR Duplikaten führen, durch technische Duplikate.
Wenn weitere bioinformatische Analysen (z. B. SNP-Identifizierung) in Auftrag gegeben werden, dann wird nur ein Exemplar des doppelten Read-Paares im Alignment behalten. Die restlichen Duplikate werden zur Vermeidung eines Bias aus der weiteren Analyse ausgenommen.
Werden alle Gene mit 10 Millionen Read-Paaren abgedeckt?
Eurofins Genomics wendet die neueste Twist Bioscience Exomanreicherung an, welches eine hervorragende einheitliche Abdeckung der Zielregionen ermöglicht. Zusätzlich ist unsere Bibliotheken-Herstellung darauf optimiert entstehenden Bias durch die Amplifikation oder das Enrichment zu minimieren.
Natürlich, gibt es aufgrund der biologischen Natur der Gene auch Regionen die schwer zu sequenzieren sind (z.Bsp hoher AT oder GC Gehalt)
Wie sieht die Qualität der Daten aus?
Bei humanen Proben, die im Paired-End-Modus (150 bp) sequenziert werden, erzielt Eurofins Genomics in der Regel über 95 % der Basecalls mit einem Qualitätswert von mehr als Q30 (> 99,9 % Genauigkeit).
Wo bekomme ich meine Ergebnisse und in welcher Form werden sie geliefert?
Die Lieferung der Alignment-Datei (BAM), der SNP- und InDel-Tabellen (VCF und TSV) sowie des Datenanalyseberichts (PDF) erfolgt in den angegebenen Formaten auf Ihr Konto.
Wie funktioniert die Datenverarbeitung mit der Ingenuity Variant Analysis von QIAGEN?
Sollten Sie sich für den optionalen Zugriff auf die Plattform der Ingenuity Variant Analysis von QIAGEN entscheiden, werden alle Rohdaten analysiert, in eine VCF-Datei konvertiert und dann direkt auf die web-basierte Software-Plattform von QIAGEN hochgeladen. Die Zugangsdaten zu Ihrem persönlichen Konto erhalten Sie von Eurofins Genomics.
Bitte beachten! Der Kunde muss den Endbenutzer-Lizenzvertrag von QIAGEN akzeptieren, bevor der Zugriff auf die Ingenuity Variant Analysis sowie alle von diesem Dienst generierten Daten gewährt wird.
Wie lange habe ich Zugriff auf meine Daten auf meinem QIAGEN-Konto?
Die Lizenz gewährt einen sechsmonatigen Zugriff auf die Analysesoftware. Nach Ablauf dieser Frist kann eine optionale Verlängerung der Lizenz ausschließlich offline erworben werden – Bitte sprechen Sie uns an!
Welche Art von Qualitätskontrollen führen Sie durch?
Qualität und Quantität jeder Probe werden bei Eingang der Probe bestimmt. Weitere Qualitätskontrollen erfolgen bei verschiedenen Schritten des Prozesses.
Welches Produkt soll ich wählen – INVIEW Core Exome (37 Mb) oder
INVIEW Whole Exome (60 Mb)?
Sowohl INVIEW Core Exome (37 Mb) als auch INVIEW Whole Exome (60 Mb) sind hervorragende Komplettlösungen für die Gesamtexom-Sequenzierung. Die Produkte bieten Sequenzierung nach Diagnosenormen (ISO 17025) sowie vergleichbare BioIT-Pipelines für die Datenanalyse. Darüber hinaus bieten beide Dienste eine ergänzende Datenverarbeitung mit der QCI Interpret Translational von QIAGEN. Die beiden Produkte unterscheiden sich geringfügig hinsichtlich ihrer Workflows, Anwendungen und den damit verbundenen Kosten.
INVIEW Core Exome (37 Mb) ist die kostengünstigste Lösung für die Gesamtexom-Sequenzierung. Das Produkt ist ideal für die Analyse von Keimbahnmutationen.
INVIEW Whole Exome (60 Mb) ist eine flexiblere Lösung für die Gesamtexom-Sequenzierung. INVIEW Whole Exome wird mit einer garantierten durchschnittlichen zielgerichteten Abdeckung in Vielfachen von 30x angeboten. Das Produkt kann zur Analyse sowohl von Keimbahnmutationen als auch von somatischen Mutationen verwendet werden.
Wohin soll ich meine Proben schicken?
Eurofins Genomics Europe Pharma and Diagnostics Products & Services Sanger/PCR GmbH
Jakob-Stadler-Platz 7
78467 Konstanz
Germany
Fragen zu INVIEW Liquid Biopsy Oncoprofiling
Welche Arten von Proben können mit INVIEW Liquid Biopsy Oncoprofiling verarbeitet werden?
Für das INVIEW Liquid Biopsy Oncoprofiling akzeptieren wir frisches Blut (muss in BCT-Röhrchen mit einem stabilisierenden, für die cfDNA-Isolation bestimmten Puffer zur Verfügung gestellt werden) oder alternativ frische oder aufbewahrte Plasmaproben, sowie bereits isolierte zellfreie DNA. Weiteres entnehmen Sie bitte unserem Informationsblatt zur Probenvorbereitung. Die cfDNA wird aus Blutplasma extrahiert.
Bei Ausgangsmaterial wie DNA, die aus FFPE-Proben und frisch eingefrorenen Gewebeproben isoliert wurde, sehen Sie sich bitte unser Produkt für die Charakterisierung fester Tumorproben an: INVIEW Oncoprofiling.
Wie viel Ausgangsmaterial wird benötigt?
Wir empfehlen die Übersendung von zwei 10 ml-Blutproben. Die erste 10 ml-Probe wird für die Tests verwendet, während die zweite Probe als Reservematerial aufbewahrt werden kann. Bei Plasmaproben ist die optimale Menge 4 ml. Bitte beachten Sie, dass die Leistung und Empfindlichkeit bei kleineren Plasmavolumen abnehmen kann. Alternativ akzeptieren wir auch 15 ng bereits isolierte zellfreie DNA(bis zu 30 µl /Konzentration > 0,5 ng/µl).
Wie erfolgt die Anreicherung der gewünschten Genabschnitte?
Die interessierenden Gene werden unter Verwendung firmeneigener Eurofins-Protokolle mit der hochmodernen Hybridisierungstechnologie SureSelect von Agilent angereichert. Das Zielregionen-Anreicherungssystem erfasst genomische Zielregionen mit Hilfe von langen 120 nt-RNA-Baits. Mit Hilfe der auf Hybridisierung basierenden Strategie können PCR-Duplikationen im Assay leicht abgezogen werden. Das Eurofins-eigene Protokoll ermöglicht die Erstellung von hochkomplexen Bibliotheken, eine tiefgreifende Abdeckung der interessierenden Regionen sowie eine verbesserte Einheitlichkeit der Sequenz. Im Anschluss an die Anreicherung der Zielregionen wird eine Next Generation Sequenzierung der Bibliothek durchgeführt.
Welche Arten von Mutationen können mit INVIEW Liquid Biopsy Oncoprofiling erkannt werden?
Das Produkt sucht die Exons von etwa 600 Genomregionen ab, die an der Entstehung von Krebs beteiligt sind. Dadurch wird eine extrem hohe Anzahl von möglichen Mutationen abgedeckt und analysiert. Zu den erkannten genomischen Aberrationen zählen SNPs, InDels, CNVs und Genfusionsereignisse.
Wie sensitiv ist das Produkt?
Die Erkennung von SNP und InDel hat eine technische Empfindlichkeit von bis zu 1 %.
Welche Art von Qualitätskontrollen führen Sie durch?
Die Qualität und Quantität jeder Probe werden bei Eingang der Probe oder nach der DNA-Extraktion bestimmt. Weitere Qualitätskontrollen erfolgen bei verschiedenen Schritten des Prozesses.
Welches Produkt soll ich wählen – INVIEW Liquid Biopsy Oncoexome, INVIEW Liquid Biopsy Oncoprofiling oder Oncotarget?
Alle drei Tests können bei Flüssigbiopsieproben (cfDNA) zum Einsatz kommen. Die drei Produkte dienen als leistungsstarkes, nichtinvasives Tool für die Krebsprofilierung.
INVIEW Liquid Biopsy Oncoexome ist der erste kommerziell erhältliche Service für die Gesamtexom-Sequenzierung (WES) von cfDNA. Mit diesem Service erhalten Sie ein umfassendes Profil aller Mutationen in den proteinkodierenden Bereichen des Exoms. Dieses Produkt eignet sich für einen eingehenden Blick auf das Exom eines Krebspatienten in Fällen, in denen nur begrenzte Informationen über klinisch relevante Mutationen zur Verfügung stehen.
INVIEW Liquid Biopsy Oncoprofiling ist ein umfassendes NGS-Krebspanel zur Charakterisierung wichtiger tumorspezifischer Mutationen, die bei fast allen Krebsarten eine Rolle spielen. Mittels SureSelect von Agilent und einem optimierten GATC-Protokoll zielt dieses Panel auf 600 bekannte Krebsgene einschließlich Tumorsuppressoren, Hotspots und Resistenz-Markern. Das Panel verfügt über eine optimierte Sequenzabdeckung und Gleichförmigkeit mit einer Sensitivität von 1 %.
INVIEW Oncotarget ermöglicht den ultrasensitiven Nachweis von klinisch relevanten krebsspezifischen Punktmutationen, Insertionen, Deletionen und Genfusionen in zirkulierender zellfreier DNA bis zu einer Allelfrequenz von 0,1 %. Diese Methode eignet sich für die Therapieüberwachung, da sie empfindlich genug ist, um ein Rezidiv in sehr frühem Stadium zu erkennen.
Kann ich INVIEW Liquid Biopsy Oncoprofiling für diagnostische Zwecke verwenden?
Das Produkt steht nur für Forschungszwecke zur Verfügung. Alle Proben werden in Erfüllung der ISO 17025:2005-Akkreditierung verarbeitet.
Wo liegen die Grenzen der CNV-Analyse?
Auch bei Einreichung von Kontrollproben (Plasmaproben von mindestens sieben andere Patienten/Personen) kann die Methode der CNV-Analyse Einschränkungen unterliegen. Sensitivität und Spezifität des Assays hängen vom Ausmaß der vorhandenen CNV sowie vom Tumoranteil ab.
Werden alle Veränderungen der Kopienzahl in meiner Probe erkannt?
Die Heterogenität eines Tumors kann eine korrekte Identifizierung aller relevanten Kopienzahlvariationen in einer gegebenen Plasmaprobe erschweren. Darüber hinaus könnte eine übermäßige Verunreinigung mit DNA aus normalen Zellen zu Unterschieden bezüglich der Abdeckung führen, die selbst bei den hochsensitiven Methoden von Eurofins Genomics zu klein sind, um nachgewiesen zu werden.
Wo bekomme ich meine Ergebnisse?
Alle Ergebnisse (FAST-Q Dateien und analysierte Daten) können über Ihren sicheren Online-Account heruntergeladen werden.
Wie schnell soll ich die Probe nach der Blutentnahme versenden?
Wie empfehlen, dass die Blutprobe innerhalb von 24 Stunden nach der Entnahme an Eurofins Genomics versandt wird. Die Probe muss in Blutentnahmeröhrchen von BCT Streck gelagert werden. Lagerung und Auslieferung von Blutproben müssen bei Raumtemperatur erfolgen. Plasmaproben müssen auf Trockeneis verschickt werden.
Wohin soll ich meine Proben schicken?
Eurofins Genomics Europe Pharma and Diagnostics Products & Services Sanger/PCR GmbH
Jakob-Stadler-Platz 7
78467 Konstanz
Germany
Fragen zu INVIEW Oncoprofiling
Welche Arten von Proben können mit INVIEW Oncoprofiling (591 genes) verarbeitet werden?
Für INVIEW Oncoprofiling (591 genes) akzeptieren wir frisch eingefrorene Proben sowie FFPE-Proben (Gewebe und Blut) und genomische DNA.
Für die Flüssigbiopsie-Analyse von aus Blutplasma isolierter zellfreier DNA (cfDNA) sehen Sie sich bitte unser Produkt INVIEW Liquid Biopsy Oncoprofiling (591 genes) an.
Wie erfolgt die Anreicherung der gewünschten Genabschnitte?
Die interessierenden Gene werden unter Verwendung firmeneigener Eurofins-Protokolle angereichert. Die Anreicherung der Zielregionen übertrifft die Ergebnisse, die auf der üblichen PCR-basierten Anreicherung erfolgen, dank der gleichmäßigen Abdeckung aller Exone der verschiedenen Gene. Die bewährte Methode zur Anreicherung von Zielregionen wurde von Eurofins optimiert: Mit ihr können hochkomplexe Bibliotheken aus geringsten DNA- Konzentrationen entwickelt werden und sie ergibt eine tiefgreifende und einheitliche Abdeckung. Im Anschluss an die Erfassung der Zielregionen wird eine Next Generation Sequenzierung der Bibliothek durchgeführt.
Welche Arten von Mutationen sind mit INVIEW Oncoprofiling (591 genes) nachweisbar?
Der Service deckt die gesamten Exons von etwa 600 krebsrelevanten Genomregionen vollständig ab, einschließlich proteinkodierender Gene, ausgewählter Promotorregionen, miRNAs, extra-exonische Varianten und ausgewählter Genfusionsereignisse. Neben SNPs und InDels kann das Produkt auch CNVs erkennen.
Wie wird der TMB (tumor mutational burden) berechnet?
- TMB is definiert als die Anzahl der somatischen, kodierenden, Basenaustausch- und InDel-Mutationen pro Megabase des kontrollierten Genoms.
Alle Basenaustauschmutationen und InDels in der kodierenden Region der Zielgene, inklusive der gleichbedeutenden Mutationen, werden zu Beginn gezählt. Anschließend werden wie unten beschrieben verschiedene Filter angewendet
- Die folgenden Mutationen werden bei der TMB Berechnung in silico ausgeschlossen:
- bekannte somatische Mutationen in COSMIC und ClinVar
- Mutationen mit geringer statistischer Sicherheit
- Bekannte Keimbahnvarianten aus der ExAC (gnomAD) Datenbank
- Mutationen die mittels dem Somatisch-Keimbahn-Zgyotie Algorithmus als somatisch eingestuft werden
- Mutationen in Tumor-Suppressor Genen aufgrund der INVIEW Oncopanel All-in-One Ausrichtung auf Krebsmutationen, die Auswirkungen haben und dem möglichen Bias des Panel-Designs
- Um den TMB pro Megabase zu berechnen wird die Gesamtzahl der gefundenen Mutationen anhand der Größe kodierenden Region in der Zielregion (in Megabasen) normalisiert (Mutationen pro Megabase, mut/Mb). Aufgrund der fehlenden Standardisierung in der TMB Quantifizierung können wir 3 Werte zur Verfügung stellen
- Nicht-gleichbedeutende Mutationen
- Nicht-gleichbedeutende Mutationen und zusätzlich InDels
- Inklusive aller Mutationen
Wie gut ist die TMB Bestimmung mit einem speziellen Panel - wie demINVIEW Oncoprofiling (591 genes) - im Vergleich zu "Whole-Exome" Sequenzierung (WES)?
Der Goldstandard für die TMB Bestimmung ist klar die "Whole-Exome" Sequenzierung (WES). Spezialisierte Panel wie das INVIEW Oncopanel All-in-One wurde aber in aufwändigen Studien auf die Verwendbarkeit für die TMB Bestimmung untersucht, da die Sequenzierkosten deutlich güngstiger und die Sensitivität deutlich höher ist.
Neueste Ergebnisse zeigen, dass Panels mit einer Größe von > 1.5 Mbp ausreichend geeignet sind um den TMB zu Bestimmung eine eine starken Übereinstimmung zu WES (Buchhalter et al. 2019, Int J Cancer 144:848–858).
Darüber hinaus liefert das INVIEW Oncopanel All-in-One 2.0 mit seiner Größe von 3 Mbp eine viel genauere TMB Bestimmung als kleinere Panels.
Wie sensitiv ist das Produkt?
Die Erkennung von SNP und InDel hat eine technische Empfindlichkeit von bis zu 1 %.
Welche Art von Qualitätskontrollen führen Sie durch?
Die Qualität und Quantität jeder Probe werden bei Eingang der Probe oder nach der DNA-Extraktion bestimmt. Weitere Qualitätskontrollen erfolgen bei verschiedenen Schritten des Prozesses.
Was ist der Unterschied zwischen INVIEW Oncoprofiling (591 genes) und INVIEW Liquid Biopsy Oncoprofiling (591 genes)?
Beide Tests bieten Variantenerkennung für Krebserkrankungen auf Basis eines validierten Onkologie-Panels. Die abgesuchten Gene und Genomveränderungen des Panels sind gleich. Ebenso basieren die verwendeten Techniken bei beiden Produkten auf Hybridisierung zur Anreicherung definierter Genabschnitte sowie auf Next Generation Sequenzierung.
Der Unterschied zwischen den beiden Produkten besteht in deren Ausgangsmaterial. INVIEW Oncoprofiling (591 genes) wird auf herkömmliches Ausgangsmaterial wie z. B. frisch eingefrorene und FFPE-Gewebeproben angewandt, während mit INVIEW Liquid Biopsy Oncoprofiling (591 genes) aus Blutplasmaproben extrahierte cfDNA analysiert wird.
Kann ich die Ergebnisse zwischen INVIEW Oncoprofiling (591 genes) und INVIEW Liquid Biopsy Oncoprofiling (591 genes) direkt vergleichen?
Ja, die beiden Dienste eignen sich bestens für Studien, deren Ziel ein Vergleich der Leistungsfähigkeit herkömmlicher Biopsien gegenüber Flüssigbiopsien ist.
Wo liegen die Grenzen der CNV-Analyse?
Auch bei Einreichung zusammengehöriger Proben (Tumorgewebe und Normalgewebe oder Blut vom selben Patienten) kann die gekoppelte Analysemethode Einschränkungen unterliegen. Die Varianz der Messungen an dieser einzelnen zugehörigen Referenzprobe wird höher sein als die einer Referenz, die aus mehreren Referenzproben besteht. Dies könnte zu einer geringen Anzahl von (falsch-positiven) CNV-Identifizierungen führen – auch in Fällen, in denen zwei zusammengehörige Proben tatsächlich die identische Kopienzahl haben.
Werden alle Varianten in meiner Probe erkannt?
Die Heterogenität eines Tumors kann eine korrekte Identifizierung aller relevanten Kopienzahlvariationen in einer gegebenen Probe erschweren. Auch wenn der Tumor selbst homogen ist, kann eine übermäßige Verunreinigung mit normalen Zellen zu Unterschieden bezüglich der Abdeckung führen, die selbst bei den hochsensitiven Methoden von Eurofins Genomics zu klein sind, um nachgewiesen zu werden.
Kann ich INVIEW Oncoprofiling (591 genes) für diagnostische Zwecke verwenden?
Das Produkt steht nur für Forschungszwecke zur Verfügung. Der Service wird in einem nach ISO17025:2005 akkreditierten und nach ISO13485:2016 zertifizierten Labor durchgeführt.
Wo erhalte ich meine Ergebnisse und in welcher Form werden sie geliefert?
Alle Ergebnisse (FAST-Q Dateien and analysierte Daten) können über Ihr sicheres Online-Account heruntergeladen werden.
Wohin soll ich meine Proben schicken?
Eurofins Genomics Europe Pharma and Diagnostics Products & Services Sanger/PCR GmbH
Jakob-Stadler-Platz 7
78467 Konstanz
Germany
Fragen zu INVIEW Metagenome Advance
Welche Art von Proben kann verwendet werden?
Für INVIEW METAGENOME ADVANCE akzeptieren wir aus der klinischen Forschung stammende Proben menschlichen Ursprungs (z. B. Abstriche, Faeces, Lavage). Auf Anfrage können für die Analyse des gesamten Metagenoms viele Arten von Ausgangsmaterialien verwendet werden, wie z. B.:
Lebensmittelproben für die Qualitätskontrolle und Nachweis von Pathogenen im industriellen Umfeld (Molkereien, Brauereien, fleischverarbeitende und landwirtschaftliche Einrichtungen)
Umweltproben
Eurofins Genomics hat besonders viel Erfahrung mit der DNA-Isolierung und Sequenzierung von Stuhlproben.
Wieviel Ausgangsmaterial wird benötigt?
Spezifische Informationen über die für spezielle Methoden der Bibliothekerstellung benötigte Menge und Konzentration finden Sie in Ihrem Angebot – oder Sie wenden sich direkt an uns. Um optimale Ergebnisse zu erzielen, benötigen wir in der Regel > 100 ng aufgereinigte Doppelstrang-DNA (die Gesamtmenge hängt vom Anteil mikrobieller DNA in der Probe ab). Bitte beachten Sie, dass die DNA RNA-frei sein und eine OD 260/280 von > 1,8 und OD 260/230 von > 2,0 haben muss.
Welche Art von Daten erhalte ich?
Eurofins Genomics liefert Tabellen (TSV, TXT) mit Auflistungen aller in Ihrer Probe vorhandenen Gattungen und den jeweiligen Read-Anzahlen. Zusätzlich erhalten Sie alle Rohdaten der Sequenzierung.
Operationale taxonomischen Einheiten (OTUs), klassifiziert vom Kraken‘schen exakten k-mer-Alignmentalgorithmus und der entsprechenden MiniKrakenDB-Datenbank, werden in Tabellenform mit taxonomischer Identifizierung dargestellt.
Welche Abdeckung ist für die Metagenom-Analyse notwendig?
Die für die Metagenom-Sequenzierung erforderliche Sequenzierungstiefe richtet sich vor allem nach der Komplexität der Probe (der Anzahl und Repräsentation der einzelnen Spezies) sowie nach dem Grad der erwarteten Wirtskontamination. Mikrobengemeinschaften hoher Komplexität mit hohen Hintergrundwerten an Wirts-DNA erfordern mehr Abdeckung als Proben geringerer Komplexität und mit einem geringeren Grad an Wirts-DNA-Kontamination. Im Zweifelsfall empfehlen wir, die erforderliche Sequenzabdeckung anhand eines Pilotversuchs an einer Untergruppe von Proben zu bestimmen.
Garantieren Sie einen bestimmten Output für die Sequenzierung?
Ja, Inview Metagenome Advance beinhaltet eine Garantie für 10 Millionen Read-Paare. Weitere Read-Pakete können separat bestellt werden.
Welche Organismen können nachgewiesen werden?
Die Metagenomanalyse ermöglicht die Identifizierung von Mikroorganismen unabhängig von taxonomischen Markern wie z. B. 16S rRNA. Anhand der Metagenomsequenzierung können sowohl kultivierbare als auch nicht-kultivierbare Organismen wie z. B. Bakterien, Archaeen, Pilze, Protozoen und Viren identifiziert werden. Zwar hat Eurofins Genomics am meisten Erfahrung mit der Profilierung der humanen Mikrobiota, wir charakterisieren jedoch auch mit Erfolg Elemente aus Mikrobengemeinschaften aus Lebensmittel-, Industrie und Umweltproben.
Welchen Service soll ich wählen – die gezielte Amplikon-Sequenzierung oder Metagenom-Sequenzierung?
Sowohl Amplikon- als auch Metagenom-Sequenzierung weisen klare Vorteile sowie auch Nachteile auf. Ihre Methode der Wahl sollte sich nach Ihrem Forschungsziel richten. Der Erfolg eines Projekts hängt in der Regel von mehreren Faktoren ab, wie z. B. von der Zusammensetzung der Gemeinschaft, der Häufigkeit eng verwandter Arten und der Sequenzierungstiefe.
Amplikon-Sequenzierung bietet eine zweckmäßige taxonomische Profilerstellung einer großen Anzahl von Proben. Dieser Ansatz ermöglicht die Erkennung von feinen Unterschieden zwischen Mikrobengemeinschaften; daher ist die Methode für statistische Vergleiche vorteilhaft, wie z. B. für Fall-Kontroll-Studien oder für Probenahmen aus unterschiedlichen Umgebungen im Lauf der Zeit.
Metagenom-Sequenzierung ist nicht auf einen bestimmten Satz von Primern angewiesen, wodurch die Methode frei ist von taxon-spezifischem PCR-Bias. Dadurch sind präzisere Darstellungen der analysierten Mikrobiota und zuverlässigere Schätzungen von Artenhäufigkeiten möglich. Die Metagenomanalyse kann Informationen sowohl über die taxonomischen als auch die funktionellen Eigenschaften einer bestimmten Probe liefern.
Was ist der Unterschied zwischen Inview Metagenome Explore und Inview Metagenome Advance?
Inview Metagenome Explore ist ideal geeignet für eine breit angelegte taxonomische Profilierung aller in einem bestimmten Mikrobiom vorhandenen Organismen. Die inbegriffenen 10 Millionen Read-Paare eignen sich zur Analyse von Metagenomen geringer Komplexität sowie auch für eine umfassende Übersicht über Gattungen in komplexen Metagenomen. Eine tiefere Sequenzabdeckung kann anhand von zusätzlichen Read-Paketen erreicht werden.
Inview Metagenome Advance bietet zusätzlich zur taxonomischen Charakterisierung eine detaillierte Beurteilung von Antibiotikaresistenzen und der Funktion in der Gemeinschaft. Das Produkt ist ideal für die Profilerstellung komplexer Metagenome. Für eine tiefere Sequenzabdeckung als die inbegriffenen 10 Millionen Read-Paare können zusätzliche Read-Pakete hinzugefügt werden. Der Service kann auf die Quantifizierung funktioneller Prozesse in einer speziellen Gemeinschaft oder auf die Erkennung zahlreicher Resistenzfaktoren in einer ungestörten natürlichen Umgebung angewendet werden. Bitte beachten Sie, dass die erforderliche Sequenziertiefe für eine erfolgreiche Metagenomprofilierung stark von der Komplexität und dem erwarteten Grad an Wirtskontamination des Ausgangsmaterials beeinflusst wird. Daher ist möglicherweise ein zusätzlicher Sequenzier-Aufwand erforderlich, um zufriedenstellende Ergebnisse zu erzielen.
Wohin soll ich meine Proben schicken?
Eurofins Genomics Europe Pharma and Diagnostics Products & Services Sanger/PCR GmbH
Jakob-Stadler-Platz 7
78467 Konstanz
Germany
Fragen zu INVIEW Metagenome Explore
Welche Art von Proben kann verwendet werden?
Generell können alle Arten von (DNA-) Proben für die Metagenom-Profilierung verwendet werden, wie z. B.:
- Proben menschlichen Ursprungs (Abstrich, Stuhl, Lavage)
- Lebensmittelproben für die Qualitätskontrolle bzgl. Mikroorganismen und den Nachweis von Pathogenen im industriellen Umfeld (Molkereien, Brauereien, fleischverarbeitende und landwirtschaftliche Einrichtungen)
- Umweltproben
Eurofins Genomics hat besonders viel Erfahrung mit der DNA-Isolierung und Sequenzierung von Stuhlproben
Wieviel Ausgangsmaterial wird benötigt?
Informationen über die für spezielle Methoden der Bibliothekerstellung benötigte Menge und Konzentration finden Sie in Ihrem Angebot – oder Sie wenden sich an uns. Um optimale Ergebnisse zu erzielen, benötigen wir in der Regel > 100 ng aufgereinigte Doppelstrang-DNA (die Gesamtmenge hängt vom Anteil mikrobieller DNA in der Probe ab). Bitte beachten Sie, dass die DNA RNA-frei sein und eine OD 260/280 von > 1,8 und OD 260/230 von > 2,0 haben muss.
Welche Art von Daten erhalte ich?
Eurofins Genomics liefert Tabellen (TSV, TXT) mit Auflistungen aller in der analysierten Probe vorhandenen Gattungen und den jeweiligen Read-Anzahlen. Zusätzlich erhalten Sie alle Rohdaten der Sequenzierung.
Operative taxonomische Einheiten (OTUs), klassifiziert durch BLAST-Analyse gegen geeignete Datenbanken, werden in tabellarischer Form einschließlich taxonomischer Identifizierung dargestellt. Für die OTU-Zuordnung werden nur die besten Treffer berücksichtigt.
Welche Abdeckung ist für die Metagenom-Analyse notwendig?
Die für die Metagenom-Sequenzierung erforderliche Sequenzierungstiefe richtet sich vor allem nach der Komplexität der Probe (der Anzahl und Repräsentation der einzelnen Spezies) sowie nach dem Grad der erwarteten Wirtskontamination. Proben hoher Komplexität mit hohen Hintergrundwerten an Wirts-DNA erfordern mehr Abdeckung als Proben geringerer Komplexität und mit einem geringeren Grad an Wirts-DNA-Kontamination. Im Zweifelsfall empfehlen wir, die erforderliche Sequenzabdeckung anhand eines Pilotversuchs an einer Untergruppe von Proben zu bestimmen.
Garantieren Sie eine bestimmten Output für die Sequenzierung?
Ja, INVIEW METAGENOME EXPLORE beinhaltet eine Garantie für 10 Millionen Read-Paare. Weitere Read-Pakete können separat bestellt werden.
Welche Organismen können nachgewiesen werden?
Die Metagenomanalyse ermöglicht die Identifizierung von Mikroorganismen unabhängig von taxonomischen genetischen Markern. Die Methode ermöglicht die Identifizierung kultivierbarer und nicht-kultivierbarer Organismen, wie z. B. Bakterien, Archaeen und Viren, sowie einfacher Eukaryoten einschließlich Pilzen und Protisten. Zwar hat Eurofins Genomics am meisten Erfahrung mit der Profilierung der humanen Mikrobiota, wir identifizieren jedoch auch mit Erfolg Elemente aus Mikrobengemeinschaften in Lebensmittel-, Industrie und Umweltproben.
Welchen Service soll ich wählen – die gezielte Amplikon-Sequenzierung oder Metagenom-Sequenzierung?
Sowohl Amplikon- als auch Metagenom-Sequenzierung haben jeweils Vorteile und Nachteile.
Ihre Methode der Wahl sollte sich nach Ihrem Forschungsziel richten. Der Erfolg eines Projekts hängt ganz allgemein von mehreren Faktoren ab, wie z. B. von der Zusammensetzung der Gemeinschaft, der Häufigkeit eng verwandter Arten oder Stämme und der Tiefe der Sequenzierungsabdeckung.
Amplikon-Sequenzierung bietet eine zweckmäßige Beurteilung der taxonomischen Zusammensetzung und Vielfalt einer großen Anzahl von Proben. Da man bessere Möglichkeiten hat, feine Unterschiede zwischen Mikrobengemeinschaften zu erkennen, ist die Methode für statistische Vergleiche vorteilhaft, wie z. B. für Fall-Kontroll-Studien oder für Probenahmen aus unterschiedlichen Umgebungen im Lauf der Zeit.
Metagenom-Sequenzierung ist aufgrund der verwendeten Zusammenstellung an Primern frei von taxonspezifischem PCR-Bias. Dies ermöglicht präzisere Darstellungen der analysierten Mikrobiota und zuverlässigere Schätzungen von Artenhäufigkeiten. Darüber hinaus können anhand der Metagenomanalyse sowohl taxonomische als auch funktionelle Eigenschaften einer bestimmten Probe aufgeklärt werden.
Was ist der Unterschied zwischen Inview Metagenome Explore und Inview Metagenome Advance?
Inview Metagenome Explore ist das ideale Produkt für die allgemeine taxonomische Profilierung aller in einem bestimmten Mikrobiom vorhandenen Organismen. Die inbegriffenen 10 Millionen Read-Paare eignen sich zur Analyse von Metagenomen geringer Komplexität sowie auch für eine umfassende Übersicht über Gattungen in komplexen Metagenomen. Eine tiefere Sequenzabdeckung kann anhand von zusätzlichen Read-Paketen erreicht werden.
Inview Metagenome Advance bietet zusätzlich zur taxonomischen Charakterisierung eine detaillierte Beurteilung von Antibiotikaresistenzen und der Funktion in der Gemeinschaft. Das Produkt ist ideal für die Profilerstellung komplexer Metagenome. Für eine tiefere Sequenzabdeckung als die inbegriffenen 10 Millionen Read-Paare können zusätzliche Read-Pakete hinzugefügt werden. Der Service unterstützt bei der Quantifizierung funktioneller Prozesse in einer speziellen Gemeinschaft oder bei der Erkennung zahlreicher Resistenzfaktoren in einer ungestörten natürlichen Umgebung.
Wohin soll ich meine Proben schicken?
Eurofins Genomics Europe Pharma and Diagnostics Products & Services Sanger/PCR GmbH
Jakob-Stadler-Platz 7
78467 Konstanz
Germany
Fragen zu INVIEW Transcriptome (Illumina)
Welches INVIEW Transkriptom-Paket empfehlen Sie für welchen Anwendungsbereich?
Dies hängt von vielen Faktoren ab, wie u. a. von der Art der Gewebes, der Probenqualität, dem Entwicklungsstadium, den vorhandenen Sequenzen usw.. Entnehmen Sie bitte der Tabelle unter „Produktspezifikationen“ Ihre optimale INVIEW Transcriptome-Lösung. Die dort angegebenen geschätzten Read-Anzahlen beziehen sich auf humane Proben. Alternativ können Sie uns kontaktieren, um Ihr Projekt mit uns zu besprechen. In bestimmten Fällen ist es möglicherweise ratsam, mit Hilfe eines Testlaufs zu ermitteln, wie viele Reads für Ihren speziellen Zweck oder Organismus benötigt werden.
Das INVIEW Transcriptome Discover verwendet eine strangspezifische RNA-Library kombiniert mit 150 bp Paired-End-Sequenzierung von Illumina. Somit empfiehlt sich das Paket für die Erkennung von differenziell exprimierten Genen, seltenen und neuen Transkripten sowie für die Entdeckung von Spleißvarianten. Darüber hinaus ermöglichen die Informationen über die Orientierung des Transkripts eine präzisere Bestimmung von Struktur und Genexpression.
Das INVIEW Transcriptome Bacteria beinhaltet 10 Millionen Reads / Read Paare und ist somit günstiger als das INVIEW Transcriptome Explore / Discover. 10 Millionen Reads / Read Paare sind aber für die meisten Untersuchungen bakterieller RNA völlig ausreichend.
Das INVIEW Transcriptome Ultra Low ist speziell für Projekte mit nur sehr sehr wenig Ausgangsmaterial entwickelt.
Brauche ich eine genomische Referenzsequenz?
INVIEW Transcriptome Discover:
Nicht unbedingt. Sollten Sie jedoch eine Referenz haben, nehmen Sie bitte die folgenden Empfehlungen unten zur Kenntnis.
INVIEW Transcriptome Bacteria / Ultra Low:
Ein klar definierter Ensembl-Name (z. B. GRCh37 oder Rnor_5.0) für die annotierte genomische Referenzsequenz muss vor Projektbeginn zur Verfügung gestellt werden. Alternativ kann die Genomsequenz in Fasta bereitgestellt werden (zusammen mit der entsprechenden Annotation im Gene Transfer Format (GTF) oder im GenBank-Format, einschließlich Annotation).
Welche Ausgangsmaterialien werden akzeptiert?
Zur Erstellung von Libraries für die Sequenzierung kann Gesamt-RNA aus Gewebe, Zellen oder Körperflüssigkeiten von eukaryotischen oder prokaryotischen Organismen verwendet werden. Zusätzlich zu aus frischem Gewebe isolierter RNA kann auch RNA aus FFPE-Proben analysiert werden. RNA-Isolation kann als Zusatzleistung angeboten werden. Bei Fragen zu Ausgangsmaterial aus anderen Quellen sprechen Sie uns bitte an.
Für das INVIEW Transcriptome Ultra Low können wir keine Zellen in Zellkultur annehmen. Desweiteren können wir hier nicht garantieren, das genug RNA extrahiert werden kann.
Wieviel Ausgangsmaterial wird benötigt?
Informationen über die für spezielle Methoden der Library-Erstellung benötigte RNA-Menge und -Konzentration finden Sie in Ihrem Angebot – oder Sie wenden sich an uns. In der Regel benötigen wir für optimale Ergebnisse 150 ng hochwertige RNA (bis zu 15 μl / optimale Konzentration 6 - 50 ng / μl (maximale Konzentration 200 ng / μl) mit einer OD 260/280 von 1,8 – 2,0 und einer OD 260/230 von 2,0 – 2,2. Möglicherweise kann mit weniger Material begonnen werden, doch dies hängt in hohem Maß von der Qualität des Materials und der gewünschten Library ab.
Die Qualität und Quantität des bereitgestellten Ausgangsmaterials sind für den Erfolg der Probenvorbereitung entscheidend. Bei Verwendung von zu wenig oder degradiertem, kontaminiertem oder anderweitig geschädigtem Ausgangsmaterial kann die Folge ein sehr niedriger Ertrag, eine fehlgeschlagene Probenvorbereitung sowie eine beeinträchtigte Qualität und Quantität der Sequenzierungsergebnisse sein.
Für das INVIEW Transcriptome Ultra Low:
Mindestens 0.15 ng/µl (in min. 10 µl). RIN > 6.5
Kann eine RNA-Isolation angeboten werden?
Eurofins Genomics bietet RNA-Isolation als Zusatzleistung an. Wir haben die Protokolle zur Extraktion von qualitativ hochwertiger RNA aus mehreren Probenarten und Ausgangsmaterialien erfolgreich optimiert. Weitere Informationen zur RNA-Isolation finden Sie in den Produktspezifikationen – oder lassen Sie sich von uns direkt beraten: Wir besprechen mit Ihnen mögliche Methoden der Probenvorbereitung für Ihre individuellen Anforderungen.
Welche Ausgangsmaterialien und Ziele sind zur Durchführung der rRNA-Abreicherung am besten geeignet?
a) Für Eukaryoten: Eine rRNA-Abreicherung empfiehlt sich für teilweise degradierte RNA oder bei Sequenzierung der RNA ohne Poly-A-Schwanz.
b) Für Prokaryoten empfehlen wir generell eine rRNA-Abreicherung, da eine poly-(A)-Anreicherung von prokaryotischen Transkripten nicht durchführbar ist.
Eine rRNA-Abreicherung kann bei Eurofins Genomics angefordert werden. Alternativ können Sie uns eine bereits rRNA-abgereicherte/mRNA-angereicherte RNA als Ausgangsmaterial zur Verfügung stellen (20 ng pro Probe (bis zu 15 µl / Konzentration >1,4 ng/µl)). Weitere Informationen finden Sie in den Produktspezifikationen
Wie effizient ist die rRNA-Abreicherung?
Die Effizienz der RNA-Entfernung richtet sich nach dem Organismus, der Zusammensetzung und der Qualität Ihrer Proben-RNA. Bei stark degradierter RNA kann eine Ribo-Abreicherung weniger effizient sein.
Soll ich mein Ausgangsmaterial auf Trockeneis verschicken oder ist Raumtemperatur ausreichend?
RNA muss auf Trockeneis versandt werden, sofern sie nicht mit Ethanol ausgefällt wurde. Gewebe/Zellkulturen müssen in flüssigem Stickstoff oder Trockeneis schockgefroren und auf Trockeneis versandt werden. Alternativ kann frisches Material in „RNAlater“ (z. B. von Ambion, SIGMA oder QIAGEN) stabilisiert und bei Raumtemperatur versandt werden. Bitte prüfen Sie im Voraus, ob der von Ihnen beauftragte Kurier Trockeneisversand anbietet.
Was soll ich tun, wenn meine Probe die Eingangs-QC nicht besteht?
Sollten die Menge, Konzentration oder Qualität des Ausgangsmaterials die Voraussetzungen für die Weiterverarbeitung nicht erfüllen, werden wir Sie kontaktieren und mit ihnen das weitere Vorgehen besprechen. Sofern möglich, wird Eurofins Genomics dann zusätzliche Vorverarbeitungsschritte zur Optimierung der Probenqualität empfehlen.
Wo und in welcher Form erhalte ich meine Ergebnisse?
Alle Rohdaten (FAST-Q Dateien) sowie die analysierten Daten können über Ihr sicheres Online-Konto heruntergeladen werden.
Welche Art von BioIT bieten Sie an?
Eine kostengünstige, hochwertige bioinformatische Analyse einschließlich Datenfilterung und QK ist optional für jedes Paket erhältlich.
Wohin soll ich meine Proben schicken?
Eurofins Genomics Europe Pharma and Diagnostics Products & Services Sanger/PCR GmbH
Jakob-Stadler-Platz 7
78467 Konstanz
Germany
Wie soll ich meine Proben verschicken?
Bitte senden Sie aufgereinigte DNA/RNA bzw. Amplikons in 1,5 ml SnapCap Tubes (ohne Parafilm). Und kleben Sie den kostenfreien NGS Barcode quer auf das Tube.
Ab 92 Proben können Sie diese auch gerne in Platten schicken. Bitte verwenden Sie hierfür "Full-Skirted" PCR Platten (zBsp von Eppendorf oder Twin).
Für mehr Informationen und den Probenversand für Extraktionsproben lesen Sie auch unseren Sample Submission Guide.
(Im Bild werden Prepaid Sanger Barcodes verwendet, aber der Ablauf ist für die kostenfreien NGS Barcodes (blau) derselbe)
|
1
Only use 1.5 ml
safe-lock tubes
|
2
Remove barcode
stricker from film
|
3
Affix barcode sticker
horizontally
|
4
QR-code visible at front so
it can be read by scaner
|
Fragen zu NGSelect Amplicon
Welches Ausgangsmaterial wird akzeptiert?
Eine Liste der akzeptierten Ausgangsmaterialien einschließlich der jeweiligen Mengen finden Sie in Tabelle 1 unter Produktspezifikationen.
Welche Art von Qualitätskontrolle führen Sie durch?
NGSelect Amplicon auf dem NovaSeq:
Vor der Sequenzierung werden Qualität und Quantität jeder Probe anhand von etablierten Methoden bestimmt (z. B. Agarosegel-Analyse / Qubit® Fluorometer / NanoDrop / Agilent 2100 Bioanalyzer). Weitere Qualitätskontrollen erfolgen bei verschiedenen Schritten des Sequenzierungsablaufs.
NGSelect Amplicon auf dem MiSeq:
Neben der Evaluation des Gelbildes, das Sie senden, dient der Erfolg der PCR-Reaktion als Qualitätskontrolle. Im Anschluss an die Bibliothekenherstellung, und an weiteren Schritten des Sequenzierungsablaufs werden weitere Qualitätskontrollen durchgeführt.
Welche Art von BioIT bieten Sie an?
Eine kostengünstige, hochwertige bioinformatische Analyse ist für jedes Paket erhältlich (weitere Informationen siehe „Produktspezifikationen“).
Kann ich die Ergebnisse für diagnostische Zwecke verwenden?
Ja. Auf Wunsch kann der Service unter diagnostischen Bedingungen mit ISO 17025-zertifizierten Prozessen durchgeführt werden.
Wo erhalte ich meine Ergebnisse und in welcher Form werden sie geliefert?
Wir stellen Ihnen die Ergebnisse über Ihren Ecom Account zu Verfügung.
Wohin soll ich meine Proben schicken?
Eurofins Genomics Europe Pharma and Diagnostics Products & Services Sanger/PCR GmbH
Jakob-Stadler-Platz 7
78467 Konstanz
Germany
Können wir für das Produkt NGSelect Amplicon auf dem MiSeq Amplikonproben einschicken, die aus mehreren verschiedenen Amplikons, generiert mit verschiedenen Primerpaaren bestehen?
Normalerweise würden Sie pro Probe 1 Amplikon senden, das mit 1 Primerpaar erzeugt wurde.
In einigen Fällen können jedoch weniger Reads pro Amplikon erforderlich sein, wie in der Projektspezifikation angegeben. In solchen Fällen können die einzelnen Amplikonproben, die Sie senden, auch aus mehreren Amplikons bestehen, die mit verschiedenen Primerpaaren generiert wurden. In diesem Fall werden wir jedoch keine Primersequenzen clippen.
Wie soll ich meine Proben verschicken?
Bitte senden Sie aufgereinigte DNA/RNA bzw. Amplikons in 1,5 ml SnapCap Tubes (ohne Parafilm). Und kleben Sie den kostenfreien NGS Barcode quer auf das Tube.
Ab 92 Proben können Sie diese auch gerne in Platten schicken. Bitte verwenden Sie hierfür "Full-Skirted" PCR Platten (zBsp von Eppendorf oder Twin).
Für mehr Informationen und den Probenversand für Extraktionsproben lesen Sie auch unseren Sample Submission Guide.
(Im Bild werden Prepaid Sanger Barcodes verwendet, aber der Ablauf ist für die kostenfreien NGS Barcodes (blau) derselbe)
|
1
Only use 1.5 ml
safe-lock tubes
|
2
Remove barcode
stricker from film
|
3
Affix barcode sticker
horizontally
|
4
QR-code visible at front so
it can be read by scaner
|
Fragen zu NGSelect DNA
Welches Ausgangsmaterial wird akzeptiert?
Eine Liste der akzeptierten Ausgangsmaterialien einschließlich der jeweiligen Mengen finden Sie in Tabelle 1 unter Produktspezifikationen.
Welche Art von Qualitätskontrolle führen Sie durch?
Qualität und Quantität jeder eingehenden Probe werden mit entsprechenden Methoden bestimmt (z. B. Agarosegel-Analyse / Qubit® Fluorometer / NanoDrop / Agilent 2100 Bioanalyzer). Weitere Qualitätskontrollen erfolgen bei verschiedenen Schritten des Prozesses.
Was soll ich tun, wenn meine Probe die Eingangs-QK nicht besteht?
Sollten die Menge, Konzentration und/oder Qualität des Ausgangsmaterials die Voraussetzungen für die Weiterverarbeitung nicht erfüllen, werden wir Sie kontaktieren und mit ihnen das weitere Vorgehen besprechen. Sofern möglich, wird Eurofins Genomics dann zusätzliche Vorverarbeitungsschritte zur Optimierung der Probenqualität empfehlen.
Welche Art von BioIT bieten Sie an?
Eine kostengünstige, hochwertige bioinformatische Analyse einschließlich Datenfilterung und QK ist für jedes Paket erhältlich.
Kann ich die Ergebnisse für diagnostische Zwecke verwenden?
Ja, auf Wunsch kann der Service unter diagnostischen Bedingungen mit ISO 17025-zertifizierten Arbeitsabläufen durchgeführt werden.
Wo erhalte ich meine Ergebnisse und in welcher Form werden sie geliefert?
Alle Rohdaten (FAST-Q Dateien) sowie die analysierten Daten können über Ihr sicheres Online-Konto heruntergeladen werden.
Wohin soll ich meine Proben schicken?
Eurofins Genomics Europe Pharma and Diagnostics Products & Services Sanger/PCR GmbH
Jakob-Stadler-Platz 7
78467 Konstanz
Germany
Wie soll ich meine Proben verschicken?
Bitte senden Sie aufgereinigte DNA/RNA bzw. Amplikons in 1,5 ml SnapCap Tubes (ohne Parafilm). Und kleben Sie den kostenfreien NGS Barcode quer auf das Tube.
Ab 92 Proben können Sie diese auch gerne in Platten schicken. Bitte verwenden Sie hierfür "Full-Skirted" PCR Platten (zBsp von Eppendorf oder Twin).
Für mehr Informationen und den Probenversand für Extraktionsproben lesen Sie auch unseren Sample Submission Guide.
(Im Bild werden Prepaid Sanger Barcodes verwendet, aber der Ablauf ist für die kostenfreien NGS Barcodes (blau) derselbe)
|
1
Only use 1.5 ml
safe-lock tubes
|
2
Remove barcode
stricker from film
|
3
Affix barcode sticker
horizontally
|
4
QR-code visible at front so
it can be read by scaner
|
Fragen zu NGSelect RNA
Welches Ausgangsmaterial wird akzeptiert?
Eine Liste der akzeptierten Ausgangsmaterialien einschließlich der jeweiligen Mengen finden Sie in Tabelle 1 unter Produktspezifikationen.
Brauche ich immer eine genomische Referenzsequenz?
In der Regel muss vor Projektbeginn ein klar definierter Ensembl-Name für die annotierte genomische Referenzsequenz zur Verfügung gestellt werden. Alternativ kann die Genomsequenz im Fasta-Format (zusammen mit der entsprechenden Annotation im Gene Transfer Format (GTF)) oder im GenBank-Format (einschließlich Annotation) bereitgestellt werden.
Welche Art von führen Sie durch?
Qualität und Quantität jeder Probe werden mit entsprechenden Methoden bestimmt (z. B. Agarosegel-Analyse / Qubit® Fluorometer / NanoDrop / Agilent 2100 Bioanalyzer). Weitere Qualitätskontrollen erfolgen bei nahezu allen Schritten des Prozesses.
Was soll ich tun, wenn meine Probe die Eingangs-QK nicht besteht?
Sollten die Menge, Konzentration oder Qualität des Ausgangsmaterials die Voraussetzungen für die Weiterverarbeitung der Probe nicht erfüllen, werden wir Sie kontaktieren und mit ihnen das weitere Vorgehen für das Projekt besprechen. Sofern möglich wird Eurofins Genomics Ihnen dann Empfehlungen zur Optimierung der Probenqualität geben.
Welche Art von BioIT bieten Sie an?
Eine kostengünstige, hochwertige bioinformatische Analyse einschließlich Datenfilterung und QK ist für jedes Paket erhältlich.
Kann ich die Ergebnisse für diagnostische Zwecke verwenden?
Ja, auf Wunsch kann der Service unter diagnostischen Bedingungen mit ISO 17025-zertifizierten Arbeitsabläufen durchgeführt werden.
Wo erhalte ich meine Ergebnisse und in welcher Form werden sie geliefert?
Alle Rohdaten (FAST-Q Dateien) sowie die analysierten Daten können über Ihr sicheres Online-Konto heruntergeladen werden.
Wohin soll ich meine Proben schicken?
Eurofins Genomics Europe Pharma and Diagnostics Products & Services Sanger/PCR GmbH
Jakob-Stadler-Platz 7
78467 Konstanz
Germany
Wie soll ich meine Proben verschicken?
Bitte senden Sie aufgereinigte DNA/RNA bzw. Amplikons in 1,5 ml SnapCap Tubes (ohne Parafilm). Und kleben Sie den kostenfreien NGS Barcode quer auf das Tube.
Ab 92 Proben können Sie diese auch gerne in Platten schicken. Bitte verwenden Sie hierfür "Full-Skirted" PCR Platten (zBsp von Eppendorf oder Twin).
Für mehr Informationen und den Probenversand für Extraktionsproben lesen Sie auch unseren Sample Submission Guide.
(Im Bild werden Prepaid Sanger Barcodes verwendet, aber der Ablauf ist für die kostenfreien NGS Barcodes (blau) derselbe)
|
1
Only use 1.5 ml
safe-lock tubes
|
2
Remove barcode
stricker from film
|
3
Affix barcode sticker
horizontally
|
4
QR-code visible at front so
it can be read by scaner
|
Fragen zu NGSelect Ready-To-Load
Welches Ausgangsmaterial wird akzeptiert?
Eine Liste der akzeptierten Ausgangsmaterialien einschließlich der jeweiligen Mengen finden Sie in Tabelle 1 unter Produktspezifikationen.
Welche Art von Qualitätskontrolle führen Sie durch?
Alle eingegangenen Ready-to-Load-Bibliotheken werden einer Qualitäts- und Quantitätskontrolle unterzogen. So wird die passende Sequenzierungsverdünnung bestimmt und es werden optimale Cluster-Dichten erzielt.
Was soll ich tun, wenn meine Probe die Eingangs-QK nicht besteht?
Sollten die Menge, Konzentration oder Qualität der sequenzierbereiten Bibliothek die Voraussetzungen für die Weiterverarbeitung der Probe nicht erfüllen, werden wir Sie kontaktieren und mit ihnen das weitere Vorgehen für das Projekt besprechen. Sofern möglich wird Eurofins Genomics Ihnen dann Empfehlungen zur Optimierung der Probenqualität geben.
Welche Art von BioIT bieten Sie an?
Eine kostengünstige und professionelle bioinformatische Analyse steht auf Anfrage zur Verfügung. Weitere Informationen finden Sie unter „Produktspezifikationen“.
Kann ich die Ergebnisse für diagnostische Zwecke verwenden?
Ja, auf Wunsch kann der Sequenzierdienst unter diagnostischen Bedingungen mit ISO 17025-Akkreditierung durchgeführt werden.
Wo erhalte ich meine Ergebnisse und in welcher Form werden sie geliefert?
Alle Rohdaten (FAST-Q Dateien) sowie die analysierten Daten können über Ihr sicheres Online-Konto heruntergeladen werden.
Garantieren Sie eine bestimmte Anzahl an Reads?
Die tatsächliche erzielte Sequenzierungsleistung (Read-Länge, Read-Qualität und Anzahl der Reads) korreliert direkt mit der Qualität der verwendeten Sequenzierungsbibliothek. GATC Biotech kann keine Garantien für Sequenzierungsdaten aus kundenseitig erstellten Ready-to-Load-Bibliotheken geben. Sollte es aufgrund von technischen Problemen mit den Sequenzierungskits oder -geräten zu unbefriedigenden Sequenzierungsergebnissen kommen, wird die Sequenzierung ohne zusätzliche Kosten für den Kunden wiederholt.
Ist das Sortieren von gemultiplexten Sequenzen inbegriffen?
Das Sortieren von Rohdaten gemäß Illumina Index Read ist inbegriffen. Der verwendete Index-Typ (Single Index oder Dual Index) sowie die entsprechenden Index-Sequenzen müssen vor dem Projektbeginn zur Verfügung gestellt werden.
Die Sortierung der Rohdaten nach kundenspezifischen Markierungen (Tags) zu Beginn des Reads kann gegen Aufpreis angeboten werden. Wir empfehlen dringend, die „lineare“ Barcode-Strategie zu vermeiden und stattdessen das Indexsystem von Illumina zu verwenden (d. h. die Barcodes werden in einem separaten Read gelesen und die Registrierung von Clustern wird nicht beeinträchtigt). Es ist unbedingt auf eine ausgewogene Basenzusammensetzung der Indizes zu achten, damit die Bildanalyse-Software die einzelnen Signale optimal unterscheiden kann.
Welche Schritte werden unternommen, um die Sequenzierungsqualität zu erhöhen?
Die Cluster-Erkennungsalgorithmen von Illumina sind so optimiert, dass die A-, T-, G- und C-Nukleotide gleichmäßig repräsentiert sind. Jede Abweichung von einer gleichmäßigen Verteilung der Basen wirkt sich negativ auf die Menge und die Qualität der erzeugten Sequenzierungsdaten aus. Bei Proben mit geringer Vielfalt oder unausgewogener Basenzusammensetzung (z. B. Amplikons, bisulfitkonvertierte Proben) wird zur Verbesserung des Nukleotid-Gleichgewichts der Bibliothek ein PhiX-„Spike-in“ (20%) eingesetzt. Das Ausmaß, in dem die negativen Auswirkungen der unausgewogenen Basenzusammensetzung durch das „Spike-in“ der PhiX-Kontrolle reduziert werden, ist von der einzelnen Probe und den Sequenzeigenschaften abhängig. Weitere Informationen entnehmen Sie bitte der Illumina-Website.
Wie wähle ich aus den verschiedenen NGSelect-Diensten den richtigen aus?
NGSelect gliedert sich in vier Hauptkategorien, die von Ausgangsmaterial, DNA, RNA, Amplikons oder Ready-to-Load-Bibliotheken abhängig sind. Jeder dieser Dienste bietet eine Vielzahl von Optionen, mit denen individuelle und erschwingliche NGS-Projekte entworfen werden können. Weitere Informationen finden Sie in Tabelle 2 (nachstehend):

Fragen zu INVIEW Liquid Biopsy Oncoexome
Welche Art von kann mit INVIEW Liquid Biopsy Oncoexome verarbeitet werden?
Für das INVIEW Liquid Biopsy Oncoexome akzeptieren wir frische oder aufbewahrte Plasmaproben, sowie bereits isolierte zellfreie DNA. Weiteres entnehmen Sie bitte unserem Informationsblatt zur Probenvorbereitung für Liquid Biopsy Produkte (siehe "Dateien"). Die ctDNA wird aus Blutplasma extrahiert.
Für Ausgangsmaterial wie z. B. aus FFPE und Gewebeproben isolierte DNA sehen Sie sich bitte unser Produkt INVIEW Human Exome Advance an.
Wie viel Ausgangsmaterial wird benötigt?
In der Regel benötigen wir entweder 4 ml Plasma oder 15 ng bereits isolierte zellfreie DNA(bis zu 30 µl /Konzentration > 0,5 ng/µl).
Welche der Sequenzierung vorausgehenden Leistungen stehen zur Verfügung?
Das Produkt INVIEW Liquid Biopsy Oncoexome umfasst mehrere Leistungen im Vorfeld der Sequenzierung, z. B. ctDNA-Extraktion aus Plasma und Bibliothekerstellung mit hoher Komplexität und niedriger Duplikatrate. Die gezielte Suche von Exons erfolgt mit SureSelect von Agilent, das eine überdurchschnittliche Exon-Anreicherung und gleichförmige Abdeckung bietet.
Was für eine Sequenzabdeckung soll ich verwenden?
Die erforderliche Abdeckung ist von der erforderlichen Empfindlichkeit der Variantenerkennung abhängig. Sie können die Sequenzabdeckung wählen, die Sie für Ihr individuelles Projekt und Studienziel benötigen.
Basierend auf Erfahrungen aus der Analyse zellfreier DNA von mehr als 40.000 Proben bis zum heutigen Tag empfehlen wir generell eine Abdeckung von 100x.
Welche Art von Qualitätskontrollen führen Sie durch?
Qualität und Quantität jeder Probe werden nach Eingang der Probe bestimmt. Weitere Qualitätskontrollen erfolgen bei verschiedenen Schritten des Prozesses.
Welches Produkt soll ich wählen – Oncoexome, Oncoprofiling oder Oncotarget?
Alle drei Tests beruhen auf Flüssigbiopsie. Bei allen drei Produkten handelt es sich um leistungsstarke, nichtinvasive Hilfsmittel zur Charakterisierung von Tumoren in Echtzeit. Mit ihrer Hilfe kann die gesamte Heterogenität der Erkrankung erfasst werden.
INVIEW Liquid Biopsy Oncoexome ist der erste kommerziell erhältliche Service für die Gesamtexom-Sequenzierung (WES) von ctDNA. Mit diesem Service erhalten Sie ein umfassendes Profil aller Mutationen in den proteinkodierenden Bereichen des Genoms. Dieses Produkt eignet sich für einen ersten Blick auf das Genom eines mutmaßlichen Krebspatienten in Fällen, in denen nur begrenzte Informationen über klinisch relevante Mutationen zur Verfügung stehen.
INVIEW Liquid Biopsy Oncoprofiling (591 genes) ist ein umfassendes NGS-Panel zur Charakterisierung wichtiger Krebsmutationen. Mittels Einzelmolekül-PCR zielt dieses Panel auf 50 bekannte Krebsgene einschließlich Tumorsuppressoren, Hotspots und Resistenz-Markern. Hinsichtlich der Sequenzabdeckung und Gleichförmigkeit mit einer Sensitivität von ca. 1 % setzt das Panel einen neuen Standard in der Branche.
INVIEW Liquid Biopsy Oncotarget ermöglicht den ultrasensitiven Nachweis von klinisch relevanten krebsspezifischen Punktmutationen, Insertionen, Deletionen und Genfusionen in zirkulierender Tumor-DNA bis zu einer Allelfrequenz von 0,1 %. Diese Methode eignet sich für die Therapieüberwachung, da sie empfindlich genug ist, um ein Rezidiv in sehr frühem Stadium zu erkennen.
Wie erhalte ich meine Ergebnisse und in welcher Form werden sie geliefert?
Alle Ergebnisse (FAST-Q Dateien) können über Ihr sicheres Online-Account heruntergeladen werden.
Wie schnell soll ich die Probe nach der Blutentnahme versenden?
Blutproben müssen innerhalb von 24 Stunden nach der Blutentnahme an Eurofins Genomics versandt werden. Die Probe muss in Blutentnahmeröhrchen von BCT Streck gelagert werden. Die Proben können bei Raumtemperatur gelagert und geliefert werden. Plasmaproben müssen auf Trockeneis verschickt werden.
Wie viele Proben kann ich schicken?
Eurofins Genomics nimmt eine beliebige Anzahl von Proben in Vielfachen von vier an.
Wohin soll ich meine Proben schicken?
Eurofins Genomics Europe Pharma and Diagnostics Products & Services Sanger/PCR GmbH
Jakob-Stadler-Platz 7
78467 Konstanz
Germany
Fragen zu INVIEW Microbiome Profiling 3.0
Welche Spezies können mit Hilfe der Mikrobiom-Analyse auf dem MiSeq detektiert werden?
Es können alle Bakterien, Archaeen und Pilze identifiziert werden, zu denen eine Sequenz in der NCBI Nukleotid Datenbank publiziert ist. Es gibt momentan zum Beispiel tausende bakterielle Spezies mit Sequenz-Einträgen in der Nukleotid Datenbank. Nah verwandte Arten mit fast identischen DNA-Sequenzen können nicht unterschieden werden. In diesen Fällen wird nur der Genus (oder die Familie) bestimmt.
Welche Einschränkungen gibt es bei der Verwendung von NGS für die Mikrobiom-Analyse?
- Nah verwandte Spezies können aufgrund ihrer fast identischen Sequenz nicht sicher voneinander unterschieden werden.
- Sehr starkt prozessierte Proben oder Proben mit einem sehr geringen pH enthalten zum Teil degradierte DNA. Dies macht eine Speziesbestimmung sehr schwierig und in manchen Fällen unmöglich.
- Je mehr Ausgangsmaterial wir erhalten, desto höher ist die Wahrscheinlichkeit gute Sequenzierergebnisse zu erzielen.
Wie hoch ist der Anteil an falsch positiven oder falsch negativen Ergebnissen?
Bei Proben mit stark degradierter DNA kann es sein, dass keine "Speziesidentifkation" möglich ist. Um falsch posistive Ergebnisse zu vermeiden, wird immer eine Negativkontrolle (Wasser) parallel zu den eigentlichen Proben prozessiert.
Mittels NGS kann jedes DNA Amplikon, das erfolgreich amplifiziert wird auch detektiert werden. Aufgrund der hohen Sensitivität der Methode, müssen Sequenzen, die nur sehr selten in der Probe vorkommen (low coverage) oder Sequenzen mit unerwarteter Sequenzlänge in der Datenanalyse entfernt werden, um falsch positive Ergebnisse, die durch PCR oder Sequenzierfehler entstanden sind, auszuschließen. Spezies, die durch weniger als 0,5% aller Reads abgedeckt werden, werden somit nicht in den PDF Report aufgenommen.
Wie ist die Lieferzeit der Mikrobiom-Analyse?
Die Lieferzeit hängt von der Anzahl der Proben ab. Sie beginnt bei 12 Tagen für bis 192 Proben. Zusätzliche Services, wie die DNA-Extraktion oder die bioinformatische Auswertung verlängern die Lieferzeit. Detaillierte Lieferzeit finden Sie in den Paketbeschreibungen im Bestell-Prozess.
Wie sollte ich die Auswahl an Target-Regionen treffen?
Wählen Sie eines oder mehrere 16S Targets aus um die bakterielle Zusammensetzung in Ihrer Probe zu analysieren, und wählen Sie eines oder beide ITS Targets aus, um die Pilze in Ihrer Probe zu klassifizieren. Für die Analyse von Archaeen wählen Sie bitte das zugehörige 16S Target aus der Liste aus.
Keine der 16S oder ITS Primer Kombination ermöglicht die Amplifikation von allen Spezies. Es ist daher zu empfehlen, mehrere Primer Kombination zu verwenden um die komplette Mikrobiom Population zu analysieren (z.B. Ihrmark et al., 2012; Toju et al., 2012)
Welche Qualitätskontrolle führen Sie bei meinen Proben durch?
Die PCR-Produkt Generierung dient uns als Qualitätskontrolle. Die PCR Bedingungen sind optimiert und der Erfolg der Amplikongenerierung ist von der Menge an bakterieller, archaeller oder Pilz-DNA in der Probe abhängig. Falls kein oder nur ein sehr schwaches Amplikon generiert werden kann, liegt dies in den meisten Fällen an einer zu geringen DNA-Menge. Standardmäßig wiederholen wir die Amplikongenerierung einmalig mit modifizierten Bedingungen in diesen Fällen.
Was passiert wenn Sie kein oder nur ein sehr schwaches Amplikon generieren können?
Wenn Eurofins Genomics bei einigen Ihrer Proben kein Amplikon erzeugen kann, dann werden wir die Bearbeitung dieser Proben einstellen und Ihnen die nicht erbrachten Sequenzierleistungen rückvergüten. Falls ein schwaches Amplikon generiert werden kann, dann wird Eurofins Genomics dieses in den Amplikonpool für die Sequenzierung mit aufnehmen. Erfahrungsgemäß werden für solche Amplikons auch nur wenige Reads generiert. Die Interpretation dieser Reads sollte mit besonderer Vorsicht erfolgen.
Kann ich Ersatzproben schicken?
Es ist nicht möglich Ersatzproben für einen bestehenden Auftrag zu senden. Falls Sie weitere Proben schicken möchten, dann platzieren Sie bitte einen neuen Auftrag. Beide Aufträge werden unabhängig voneinander bearbeitet.
Werden meine Proben mit den Proben anderer Kunden auf einem Lauf sequenziert?
Ja. Ihre Proben werden gemeinsam mit den Proben anderer Kunden auf einem MiSeq Run sequenziert.
Werden Sie meine Proben nach der Prozessierung aufbewahren?
Ihre Proben werden unter geeigneten Bedingungen für 3 Monate nach der Analyse aufbewahrt. Im Anschluss werden die Proben entsorgt, falls keine gesonderten Absprachen vorliegen. Bitte beachten Sie, dass Eurofins Genomics möglicherweise das komplette Probenmaterial für die Analyse verwendet (abhängig davon wieviel Material Sie zu Verfügung stellen können).
Wenn wir das Microbiome Produkt - eigene Targets auswählen, können wir dann Amplikonproben einschicken, die aus mehreren verschiedenen Amplikons, generiert mit verschiedenen Primerpaaren bestehen?
Normalerweise würden Sie pro Probe 1 Amplikon senden, das mit 1 Primerpaar erzeugt wurde. In einigen Fällen können jedoch weniger Reads pro Amplikon erforderlich sein, wie in der Projektspezifikation angegeben. In solchen Fällen können die einzelnen Amplikonproben, die Sie senden, auch aus mehreren Amplikons bestehen, die mit verschiedenen Primerpaaren generiert wurden. In diesem Fall werden wir jedoch keine Primersequenzen clippen und Rohdaten versenden.
Wie soll ich meine Proben verschicken?
Bitte senden Sie aufgereinigte DNA/RNA bzw. Amplikons in 1,5 ml SnapCap Tubes (ohne Parafilm). Und kleben Sie den kostenfreien NGS Barcode quer auf das Tube.
Ab 92 Proben können Sie diese auch gerne in Platten schicken. Bitte verwenden Sie hierfür "Full-Skirted" PCR Platten (zBsp von Eppendorf oder Twin).
Für mehr Informationen und den Probenversand für Extraktionsproben lesen Sie auch unseren Sample Submission Guide.
(Im Bild werden Prepaid Sanger Barcodes verwendet, aber der Ablauf ist für die kostenfreien NGS Barcodes (blau) derselbe)
|
1
Only use 1.5 ml
safe-lock tubes
|
2
Remove barcode
stricker from film
|
3
Affix barcode sticker
horizontally
|
4
QR-code visible at front so
it can be read by scaner
|
Fragen zu INVIEW CRISPR Check
Welche Arten von Proben können mit INVIEW CRISPR Check verarbeitet werden?
The starting material is amplicons prepared in your lab that span the mutation site. The amplicon generation protocol to apply depends on the product type (adaptor ligation or 2nd PCR workflow), instructions can be found in the tab "How to prepare your Amplicons" on the respective product webpage. Amplicon length requirements can be found in the tab "Specifications".
Als Startmaterial benötigen wir von Ihnen Amplikons, welche die Mutationsstelle überspannen. Das Vorgehen zur Amplikongenerierung hängt von dem Produkttyp ab (Adaptor Ligation oder 2nd PCR Workflow). Instruktionen können Sie auf der Webseite auf dem Tab "Probenvorbereitung" finden. Anforderungen zur Amplikonlänge finden Sie auf dem Tab "Spezifikationen".
Warum empfehlen Sie eine relativ engen Bereich, innerhalb dem das Wildtyp-Amplikon liegen sollte?
Die Amplikonlänge (Wildtyp) muss an die Länge der InDels angepasst werden, die Sie detektieren möchten. Zudem muss die Amplikonlänge ein Merging überlappender Reads zulassen. Üblicherweise sind die Mutationen die Sie mit CRISPR (NHEJ Pathway) einführen im Bereich von 1-20 bp). Um auch größere InDels nicht von der Analyse auszuschließen muss die Wildtyp Amplikongrößer in einem bestimmten Größenbereich liegen.
Sie finden in unserem Sample Preparation Guide die Längenanforderungen für alle Produkttypen und für zwei Beispiel Anwendungsfälle.
Können wir Ihren Service auch nutzen wenn wir mit einem anderen CRISPR System arbeiten als dem SpCas9 System?
Ja, Sie können. In diesem Fall benötigen wir für die Analyse das Schnittstellen Offset (Distanz in Nukleotiden upstream der PAM Sequenz, bei der der Doppelstrangbruch erwartet wird).
Haben Sie Empfehlungen für das Assay Setup?
- Bitte fügen Sie immer mindestens eine Wildtyp Kontrollprobe in Ihr Experiment ein.
- Wenn Sie eine sehr akkurate Bestimmung der Editing Efficiency benötigen, dann empfehlen wir Ihnen Replikate in die Analyse aufzunehmen. Replikate erhöhen die Genauigkeit der Effizienzbestimmung, sind aber nicht zwingend notwendig.
Wie unterscheidet sich INVIEW CRISPR Check Adaptor Ligation und INVIEW CRISPR Check 2nd PCR?
Beide Produkte sind exzelleente Lösungen um Mutationen zu untersuchen die mit dem CRISPR/Cas System (NHEJ pathway) eingeführt wurden.
The Produkte unterscheiden sich vor allem in der Library Generierung (2-Step Protokoll versus Adaptor Ligations Protokoll) and damit in der Amplikon-Probenvorbereitung.
Ein Selection Guide ist auf der CRISPR Webseite verfügbar (Tab Overview) um Ihnen zu helfen die beste Lösung für Ihre individuellen Präferenzen und Gegebenheiten zu finden.
Wie sieht der Lieferumfang aus?
- Umfassender Bericht mit Abschnitten zu Ergebnissen und Methoden
- Rohdaten als FASTQ Dateien
- FASTQ Sequenzen nach Qualitäts-Clipping und Merging
- Gemappte Reads im BAM Format
- Sequenz des Wildtypallels und alternativer Allele inklusive deren Quantität
- Metriken zu Rohdaten, Präprozessierungs-Daten, Mappingergebnisse und Coverage im CSV-Format
- Berechnete Editing Effizienz pro Probe
Wie erhalte ich meine Ergebnisse?
Für das Produkt INVIEW CRISPR Check 2nd PCR können Sie die Ergebnisse von unserem sicheren FTP Server herunterladen (passwort wird Ihnen mitgeteilt). Für das Produkt INVIEW CRISPR Check Adaptor Ligation stellen wir Ihnen die Ergebnisse über Ihren Ecom Account zu Verfügung.
Wohin soll ich meine Proben schicken?
Eurofins Genomics Europe Pharma and Diagnostics Products & Services Sanger/PCR GmbH
Jakob-Stadler-Platz 7
78467 Konstanz
Germany
Können wir für das Produkt INVIEW CRISPR Check 2nd PCR Amplikonproben einschicken, die aus mehreren verschiedenen Amplikons, generiert mit verschiedenen Primerpaaren bestehen?Normalerweise würden Sie pro Probe 1 Amplikon senden, das mit 1 Primerpaar erzeugt wurde. In einigen Fällen können jedoch weniger Reads pro Amplikon erforderlich sein, wie in der Projektspezifikation angegeben. In solchen Fällen können die einzelnen Amplikonproben, die Sie senden, auch aus mehreren Amplikons bestehen, die mit verschiedenen Primerpaaren generiert wurden. In diesem Fall werden wir jedoch keine Primersequenzen clippen und Rohdaten versenden.
Wie soll ich meine Proben verschicken?
Bitte senden Sie aufgereinigte DNA/RNA bzw. Amplikons in 1,5 ml SnapCap Tubes (ohne Parafilm). Und kleben Sie den kostenfreien NGS Barcode quer auf das Tube.
Ab 92 Proben können Sie diese auch gerne in Platten schicken. Bitte verwenden Sie hierfür "Full-Skirted" PCR Platten (zBsp von Eppendorf oder Twin).
Für mehr Informationen und den Probenversand für Extraktionsproben lesen Sie auch unseren Sample Submission Guide.
(Im Bild werden Prepaid Sanger Barcodes verwendet, aber der Ablauf ist für die kostenfreien NGS Barcodes (blau) derselbe)
|
1
Only use 1.5 ml
safe-lock tubes
|
2
Remove barcode
stricker from film
|
3
Affix barcode sticker
horizontally
|
4
QR-code visible at front so
it can be read by scaner
|
Fragen zu INVIEW Virus
Brauche ich immer eine genomische Referenzsequenz?
Falls Sie Ihr Projekt mit "Varianten-Analyse" bestellen möchten, dann benötigen wir eine Referenz-Sequenz.
In der Regel muss vor Projektbeginn ein klar definierter Ensembl-Name für die annotierte genomische Referenzsequenz zur Verfügung gestellt werden. Alternativ kann die Genomsequenz im Fasta-Format (zusammen mit der entsprechenden Annotation im Gene Transfer Format (GTF)) oder im GenBank-Format (einschließlich Annotation) bereitgestellt werden.
Welche Art von führen Sie durch?
Qualität und Quantität jeder Probe werden mit entsprechenden Methoden bestimmt (z. B. Agarosegel-Analyse / Qubit® Fluorometer / NanoDrop / Agilent 2100 Bioanalyzer). Weitere Qualitätskontrollen erfolgen bei nahezu allen Schritten des Prozesses.
Was soll ich tun, wenn meine Probe die Eingangs-QK nicht besteht?
Sollten die Menge, Konzentration oder Qualität des Ausgangsmaterials die Voraussetzungen für die Weiterverarbeitung der Probe nicht erfüllen, werden wir Sie kontaktieren und mit ihnen das weitere Vorgehen für das Projekt besprechen. Sofern möglich wird Eurofins Genomics Ihnen dann Empfehlungen zur Optimierung der Probenqualität geben.
Kann ich die Ergebnisse für diagnostische Zwecke verwenden?
Ja, auf Wunsch kann der Service unter diagnostischen Bedingungen mit ISO 17025-zertifizierten Arbeitsabläufen durchgeführt werden.
Wo erhalte ich meine Ergebnisse und in welcher Form werden sie geliefert?
Alle Rohdaten (FAST-Q Dateien) sowie die analysierten Daten können über Ihr sicheres Online-Konto heruntergeladen werden.
Wohin soll ich meine Proben schicken?
Eurofins Genomics Europe Pharma and Diagnostics Products & Services Sanger/PCR GmbH
Jakob-Stadler-Platz 7
78467 Konstanz
Germany
Wie soll ich meine Proben verschicken?
Bitte senden Sie aufgereinigte DNA/RNA bzw. Amplikons in 1,5 ml SnapCap Tubes (ohne Parafilm). Und kleben Sie den kostenfreien NGS Barcode quer auf das Tube.
Ab 92 Proben können Sie diese auch gerne in Platten schicken. Bitte verwenden Sie hierfür "Full-Skirted" PCR Platten (zBsp von Eppendorf oder Twin).
Für mehr Informationen und den Probenversand für Extraktionsproben lesen Sie auch unseren Sample Submission Guide.
(Im Bild werden Prepaid Sanger Barcodes verwendet, aber der Ablauf ist für die kostenfreien NGS Barcodes (blau) derselbe)
|
1
Only use 1.5 ml
safe-lock tubes
|
2
Remove barcode
stricker from film
|
3
Affix barcode sticker
horizontally
|
4
QR-code visible at front so
it can be read by scaner
|
Fragen zu INVIEW Plasmid-Verifizierung
Welches Ausgangsmaterial wird akzeptiert?
Eine Liste der akzeptierten Ausgangsmaterialien einschließlich der jeweiligen Mengen finden Sie in Tabelle 1 unter Produktspezifikationen.
In welchem Format muss ich die Referenzsequenz zur Verfügung stellen?
Die Referenzsequenz kann im Fragebogen zu den Probeninformationen hochgeladen werden. Die Datei sollte .doc, .docx, .xls, oder .xlsx sein. Und die Datei sollte nicht größer als 2MB sein.
Wo erhalte ich meine Ergebnisse und in welcher Form werden sie geliefert?
Alle Rohdaten (FAST-Q Dateien) sowie die analysierten Daten können über Ihr sicheres Online-Konto heruntergeladen werden.
Wohin soll ich meine Proben schicken?
Eurofins Genomics Europe Pharma and Diagnostics Products & Services Sanger/PCR GmbH
Jakob-Stadler-Platz 7
78467 Konstanz
Germany
Wie soll ich meine Proben verschicken?
Bitte senden Sie aufgereinigte DNA/RNA bzw. Amplikons in 1,5 ml SnapCap Tubes (ohne Parafilm). Und kleben Sie den kostenfreien NGS Barcode quer auf das Tube.
Ab 92 Proben können Sie diese auch gerne in Platten schicken. Bitte verwenden Sie hierfür "Full-Skirted" PCR Platten (zBsp von Eppendorf oder Twin).
Für mehr Informationen und den Probenversand für Extraktionsproben lesen Sie auch unseren Sample Submission Guide.
(Im Bild werden Prepaid Sanger Barcodes verwendet, aber der Ablauf ist für die kostenfreien NGS Barcodes (blau) derselbe)
|
1
Only use 1.5 ml
safe-lock tubes
|
2
Remove barcode
stricker from film
|
3
Affix barcode sticker
horizontally
|
4
QR-code visible at front so
it can be read by scaner
|
Fragen zu Bakt-Genomsequenzierung
Kann ich meine Proben auch ohne den kostenlosen Barcode schicken?
Leider nicht. Dieser Barcode wird für unsere internen Laborprozesse benötigt und ist wesentlich, um sicherzustellen, dass wir Ihre Proben so effizient wie möglich bearbeiten können. Vielen Dank für Ihre Mitarbeit!
Bietet Eurofins Genomics eine Prepaid Lösung für die Baktierelle Genomsequenzierung an?
Tolle Neuigkeiten! Wir arbeiten aktiv daran, Ihnen eine Prepaid-Lösung zu bringen, die in Kürze verfügbar sein wird!
Was ist die Lieferzeit für die Genomsequenzierung von Bakterien?
Im Durchschnitt beträgt die Lieferzeit 5 - 7 Arbeitstage für alle Bakterien-Genome bis zu 7 Mb.
Problembehandlungshandbuch für Ihre Ergebnisse
Es gibt mehrere Gründe, warum Ihre Ergebnisse von Ihren Erwartungen abweichen.
Um Ihre Ergebnisse zu interpretieren, werfen Sie einen Blick auf unser Problembehandlungshandbuch. Klicken Sie hier >>
Wird die Sequenzierung meiner Probe wiederholt, wenn die erste Sequenzierung fehlschlägt?
Für jede Durchführung führen wir mehrere Qualitätskontrollmessungen durch, um den Erfolg der Bibliotheksvorbereitung und Sequenzierung zu bewerten. Wenn alle Messungen zufriedenstellende Ergebnisse anzeigen, werden wir Proben, die fehlgeschlagen sind, nicht erneut verarbeiten.
Mit welchem Genauigkeitsniveau kann man bei den Ergebnissen der Sequenzierung des bakteriellen Genoms rechnen?
Gemäß den Spezifikationen von Oxford Nanopore für die v14 Library-Prep-Chemie und die derzeit in der bakteriellen Genomsequenzierung verwendeten R10.4.1-Flowcells übersteigt die Rohlesen-Genauigkeit 99%. Die Genauigkeit der endgültigen Assemblierung variiert je nach Abdeckung und Datenqualität. Im Allgemeinen führt eine höhere Abdeckung, die mehr Reads für den Konsensbau bedeutet, zu einer verbesserten Ergebnisgenauigkeit.
Ist es möglich, eine Mischung von bakteriellen Arten in einer Probe zu sequenzieren?
Ist es möglich, die nativen Plasmide meiner Bakterien zusammen mit ihrer genomischen DNA (gDNA) zu sequenzieren?
Wenn Ihre genomische DNA (gDNA)-Extraktion auch native Plasmid-DNA enthält, ist es wahrscheinlich, dass Sie Sequenzierungsreads für diese Plasmide erhalten. Im Allgemeinen werden DNA-Fragmente mit einer Größe von <1 kb während der Sequenzierung ausgeschlossen. Kleine, plasmidgroße Reads werden jedoch während der Assemblierung nicht herausgefiltert, daher besteht die Wahrscheinlichkeit, dass sie zu eigenen Plasmidassemblierungen beitragen, zusätzlich zur gDNA-Assemblierung.
Letztendlich hängt die Art der in Ihrer Probe enthaltenen DNA, die zu einer Assemblierung beiträgt, von der Gesamtqualität der Probe, der Abdeckung und dem relativen Vorkommen oder der Degradation jedes DNA-Typs ab.
Können Sie auch Genome sequenzieren, die größer als 7 Mb sind oder Genome, die linear oder mehrchromosomal sind?
Natürlich. Details zu unserem gesamten NGS ONT Portfolio finden sie hier>>
Wie sollen die Fehler bewertet werden, die ich einer Homopolymeregion oder an einer Dam- oder Dcm-Methylierungsstelle entdeckt habe?
Die vorherrschenden Fehlermodi beim Oxford Nanopore-Sequenzieren umfassen Deletionen innerhalb von Homopolymer-Bereichen, Fehler, die an der zentralen Position der Dcm-Methylierungsstelle CCTGG oder CCAGG auftreten, sowie Fehler an der Dam-Methylierungsstelle GATC.
Bitte behandeln Sie diese Regionen daher besonders sorgfältig.
Fragen zu klonaler Amplikonsequenzierung
Kann ich meine Proben auch ohne den kostenlosen Barcode schicken?
Leider nicht. Dieser Barcode wird für unsere internen Laborprozesse benötigt und ist wesentlich, um sicherzustellen, dass wir Ihre Proben so effizient wie möglich bearbeiten können. Vielen Dank für Ihre Mitarbeit!
Wie sollen die Fehler bewertet werden, die ich einer Homopolymeregion oder an einer Dam- oder Dcm-Methylierungsstelle entdeckt habe?
Die vorherrschenden Fehlermodi beim Oxford Nanopore-Sequenzieren umfassen Deletionen innerhalb von Homopolymer-Bereichen, Fehler, die an der zentralen Position der Dcm-Methylierungsstelle CCTGG oder CCAGG auftreten, sowie Fehler an der Dam-Methylierungsstelle GATC.
Bitte behandeln Sie diese Regionen daher besonders sorgfältig.
Warum fehlen einige terminalen Nukleotide in meiner linearen Amplikon-Konsensussequenz?
Abhängig von der Sequenz Ihres Probenmaterials kann der Assembler gelegentlich Schwierigkeiten bei der Rekonstruktion der terminalen Enden von linearer DNA haben, was dazu führen kann, dass bis zu 10 Nukleotiden an den 3'- und/oder 5'-Enden Ihres Inserts fehlen.
Wenn Sie dieses Problem bei Ihren Proben feststellen, können Sie die Rohdaten von Ihrem Dashboard herunterladen und die Enden mit Ihrer bevorzugten Methode rekonstruieren.
Werden meine Amplikons nochmals sequenziert, wenn kein Ergebnis erzielt wird?
Für jeden Lauf führen wir mehrere Qualitätskontrollmessungen durch, um den Erfolg der Bibliotheksvorbereitung und Sequenzierung zu bewerten. Wenn alle Messungen zufriedenstellende Ergebnisse zeigen, werden wir Proben, die fehlgeschlagen sind, nicht erneut bearbeiten.
Mit welcher Sequenzabdeckung kann ich für meine klonalen Amplicons rechnen?
Wir bieten keine spezifische Abdeckungsgarantie an. Die Anzahl der generierten Rohdaten kann erheblich je nach Probenqualität variieren. Erfolgreiche Proben, die in der erforderlichen Konzentration eingesendet werden, ergeben in der Regel eine Anzahl von Rohsequenzierungsdaten im Bereich von Dutzenden bis zu Hunderten (oder sogar Tausenden).
Die durchschnittliche Abdeckung Ihrer Probe ist im Ergebnisbericht HTML enthalten, wobei eine Abdeckung von über ~20x auf einen sehr genauen Konsens hinweist.
Kann ich Mischungen von Amplicons einsenden?
Unser Amplicon-Service ist auf eine Population von Molekülen ausgelegt, die klonal ist.
Was passiert, wenn Sie Mischungen einsenden?
Wenn Ihre Arten ausreichend unterschiedlich sind (zum Beispiel in Größe oder Sequenz stark unterschiedlich), wird der Ablauf zunächst versuchen, eine .fasta-Konsensdatei aus der am häufigsten vorkommenden Art zu erstellen. Es wird auch versucht, einen Konsens für andere Arten zu erstellen, deren Lesanzahlen mehr als 20% derjenigen der am häufigsten vorkommenden Art entsprechen. Letztendlich hängt es von der Gesamtqualität der Probe, der Abdeckung und der relativen Häufigkeit/Abbau jeder Art ab, welche Arten einen Konsens erzeugen.
Wie kann ich meine Amplicon-Mischung sequenzieren lassen?
Eurofins Genomics bietet ein umfangreiches Portfolio für die Sequenzierung von Amplicons. Sie werden definitiv den passenden Service für Ihre Bedürfnisse finden: Amplicon-Portfolio >>