The right answers to frequently asked questions
Find the answers to all our products and services by clicking the links below.
Next Generation Sequencing
Are there special features regarding the standard DIN EN ISO 17025?
Under the conditions listed below, only simplified test reports may be provided.
Subject: Simplified test reports according to DIN EN ISO 170252018, (Section 7.8)
Testing Laboratory (Accreditation): Eurofins Genomics Europe Applied Genomics GmbH (D-PL-13372, ) Eurofins Genomics Europe Sequencing GmbH (D-PL-17038)
For accredited examinations at these two companies, test results may only be reported in simplified form, i. Requirements of the standard DIN EN ISO 17025: 2018 (section 7.8) are not fully applicable to this type of transfer of results.
These results can therefore also be reported as short test reports, electronically generated result reports, Excel spreadsheets or as result files.
If you have any questions, please contact the Customer Care Team.
Questions on INVIEW Resequencing
Where do I get my results?
All raw data (FAST-Q files) as well as the analysed and assembled data can be downloaded via your secure Ecom online account.
How should I send my samples?
- Please use 1.5 ml safe-lock tubes for your templates and primers
- Do not tape or wrap tubes with parafilm. Safe-lock tubes offer perfect sealing and evaporation protection
- Label your template with our free NGS barcode labels.
If you send more than 92 samples then you can also sent the samples in plates. Please use full skirted PCR plates (e.g. Eppendorf or Twin).
For more information and for sample submission for extraction please read our Sample Submission Guide.
(Picture shows the procedure with Sanger Prepaid Barcodes but work the same with free NGS Barcodes)
|
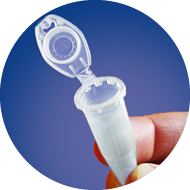
1
Only use 1.5 ml
safe-lock tubes
|

2
Remove barcode
stricker from film
|
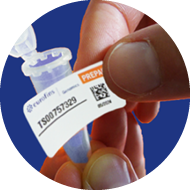
3
Affix barcode sticker
horizontally
|
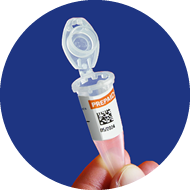
4
QR-code visible at front so
it can be read by scaner
|
Where should I send my samples?
Eurofins Genomics Europe Pharma and Diagnostics Products & Services Sanger/PCR GmbH
Jakob-Stadler-Platz 7
78467 Konstanz
Germany
Questions on INVIEW Human Exome
What is the data handling process, including QIAGEN’s QCI Interpret Translational?
All raw data are analysed and converted into a vcf file, which is uploaded directly to QIAGEN’s web-based software platform. Log-in data to your account will be provided by Eurofins Genomics.
Furthermore, the alignment file (BAM), the SNP and InDel tables (vcf and tsv) and the Data Analysis Report (pdf) from Eurofins Genomics will be delivered in the formats indicated via your online account (see below).
Please note: Customers must accept the QIAGEN End-User License Agreement before access will be granted to the Variant Analysis product or to the results generated by this product.
How long is my data accessible at my QIAGEN account?
The license grants you initial access to the analysis software for 6 months. After that period you have the option of renewing the license, but you may only purchase a renewal offline – please contact us.
Where do I get my results, if I only choose the optional BioIT?
All raw data as well as the analysed data can be downloaded via your secured online account.
What coverage should I use?
For SNP calling in heterozygote organisms we recommend at least 30x coverage. A higher coverage will increase the confidence of SNP calling. Confidently discovering genetic variants in inhomogeneous samples, such as DNA extracted from normal and tumour cells, requires higher overall coverage. The amount of data needed to reach a certain coverage on the DNA of interest depends on the ratio of normal-to-tumour DNA. Please note that due to varying efficiency of the enrichment baits, the targeted regions are not covered evenly (see below).
How do you handle PCR duplicates?
PCR duplicate rates are directly correlated to the quality and amount of starting material provided. Library preparation with less than the recommended amount of DNA requires additional PCR cycles to generate enough material to load the sequencer. Hence the PCR duplicate rate is dependent on the sample and cannot be influenced by Eurofins Genomics.
Duplicates are not excluded from the calculation of the average on-target coverage. Based on our experience with the sequencing of human samples, we typically obtain PCR duplicate rates of approximately 5% for high-quality DNA.
If further bioinformatics are ordered (e.g., SNP identification), only one copy of the duplicate read pair is kept in the alignment. The rest are excluded from further analysis to prevent any bias.
Which starting material should I send? In what quantities?
For optimum results we require at least 100 ng double-stranded, purified, high-molecular-weight, RNA-free DNA (concentration ≥ 1 ng/µL; OD 260/280 ≥ 1.8; OD 260/230 ≥ 2.0). For DNA from FFPE samples we need at least 200 ng of DNA.
Contamination by DNA from other species (> 3%; especially from closely related species) might interfere with the enrichment of the human DNA and also reduces the total amount of human DNA in the provided sample. Whole-genome amplified (WGA) DNA or DNA from archived tissue, such as Formalin-Fixed Paraffin Embedded (FFPE) samples, is usually accepted. Please contact us if you need any details.
What do you mean by overlapping reads?
The fragmentation of genomic DNA according to the Agilent SureSelectXT protocol and subsequent downstream processing produces a Gaussian distribution of DNA fragments with different lengths. A small percentage of the resulting library fragments have an insert size below 250 bp. Sequencing those library fragments with 150 bp paired-end reads will generate partially overlapping reads that cover the same bases and hence create an artificial doubling of coverage at those positions. To assure accurate analyses, those bases are excluded from further downstream analyses, such as single-nucleotide variant and insertion and deletion detection. Furthermore, Eurofins Genomics is continuously optimising its protocols to minimise the number of overlapping bases.
What is the quality of the data?
When sequencing human samples in 150 bp paired-end mode, Eurofins Genomics typically achieves over 80% of base calls with a quality value higher than Q30 ( >99.9% accurate).
Do you guarantee a specific on-target output?
INVIEW Human Exome Premium
Yes. For samples that have successfully passed the initial quality check at Eurofins Genomics, we will guarantee the on-target output you have ordered. Average on-target coverage is calculated as the sum of the mapped bases at each target position, divided by the number of bases in the target (bases on-target / size of target region).
INVIEW Human Exome Standard
No. The on target coverage cannot be guaranteed.
Which genes are enriched?
The target region corresponds to the bait coordinates of the Agilent SureSelect Human All Exon V8 Kit
Are all genes covered at 30x?
Due to varying efficiency of the enrichment baits (e.g., GC / AT content), targeted regions are covered differently and the range and uniformity of coverage varies over the target region. Eurofins Genomics applies the most recent Agilent Human All Exon kit design (V8) with improved design algorithms to better capture difficult regions and provide superior coverage uniformity.
What kind of quality controls do you perform?
The quality and quantity of each incoming sample will be determined by appropriate methods. Further quality controls are performed at various steps of the process.
Where should I send my samples?
Eurofins Genomics Europe Pharma and Diagnostics Products & Services Sanger/PCR GmbH
Jakob-Stadler-Platz 7
78467 Konstanz
Germany
How should I send my samples?
- Please use 1.5 ml safe-lock tubes for your templates and primers
- Do not tape or wrap tubes with parafilm. Safe-lock tubes offer perfect sealing and evaporation protection
- Label your template with our free NGS barcode labels.
If you send more than 92 samples then you can also sent the samples in plates. Please use full skirted PCR plates (e.g. Eppendorf or Twin).
For more information and for sample submission for extraction please read our Sample Submission Guide.
(Picture shows the procedure with Sanger Prepaid Barcodes but work the same with free NGS Barcodes)
|
1
Only use 1.5 ml
safe-lock tubes
|
2
Remove barcode
stricker from film
|
3
Affix barcode sticker
horizontally
|
4
QR-code visible at front so
it can be read by scaner
|
Questions on INVIEW Oncopanel All-in-one Liquid Biopsy
What kind of samples can be processed with INVIEW Oncopanel All-in-one Liquid Biopsy?
For INVIEW Oncopanel All-in-one Liquid Biopsy, we accept fresh blood (must be provided in BCT with a stabilising buffer intended for the isolation of cfDNA) or plasma samples or retained plasma samples as well as already isolated cell-free DNA. Please refer to our sample preparation sheet. The extraction of cfDNA is performed from the blood plasma fraction.
For starting material like DNA isolated from FFPE and fresh-frozen tissue samples, please refer to our product aimed at solid tumour profiling, INVIEW Oncopanel All-In-One.
How much starting material is required?
We recommend the delivery of two 10 ml blood samples. The first 10 ml blood sample is used for testing, whereas the second sample can be stored as reserve material. For plasma samples the optimal amount is 4 ml. Please note, that with smaller plasma volumes the performance and sensitivity can decrease. Alternatively we accept 15 ng already isolated cell-free DNA (up to 30 µl / concentration > 0,5 ng/µl).
How is the target enrichment performed?
The genes of interests are enriched via proprietary Eurofins protocols using the latest Agilent SureSelect hybridisation technology. The target enrichment system captures genomic targets using long 120 nt RNA baits. The hybridisation-based strategy facilitates the deduction of PCR duplications in the assay. The Eurofins proprietary protocol enables the generation of highly complex libraries, deep coverage of regions of interest and improved sequence uniformity. Following target enrichment, next-generation sequencing of the library is performed.
What kind of mutations can be detected with INVIEW Oncopanel All-in-one Liquid Biopsy?
The product interrogates the exons of about 600 genomic regions that are implicated in cancer development. Thereby, an extremely high number of possible mutations are covered and analysed. The detected genomic aberrations include SNPs, InDels, CNVs and gene fusion events.
What is the sensitivity of the product?
SNP and InDel detection has a technical sensitivity down to 1%.
What kind of quality controls do you perform?
The quality and quantity of each sample will be determined at sample receipt or after DNA extraction. Further quality controls are performed at various steps of the process.
How should I choose between INVIEW Oncoexome, Oncopanel All-In-One and Oncotarget Liquid Biopsy?
All three tests can be used with liquid biopsy samples (cfDNA). The three products serve as powerful non-invasive tools for cancer profiling.
INVIEW Oncoexome Liquid Biopsy is the first commercially available service for whole exome sequencing (WES) of cfDNA. With this service, a comprehensive profile of all mutations present in the protein coding regions of the exome is obtained. This product is suitable for a comprehensive look at the exome of a cancer patient, in cases where limited information regarding clinically relevant mutations is available.
INVIEW Oncopanel All-in-one Liquid Biopsy is a comprehensive NGS cancer panel for profiling important tumour-specific mutations implicated in nearly all cancer types. The service uses Agilent SureSelect with an optimized protocol to target about 600 known cancer genes including tumour suppressors, mutation hotspots and drug resistance markers. The panel is optimised for sequence coverage and uniformity with sensitivity down to 1%.
INVIEW Oncotarget facilitates ultra-sensitive detection of clinically relevant cancer-specific point mutations, insertions, deletions and gene fusions in circulating cell-free DNA down to an allele frequency of 0.1%. This approach is suitable for treatment monitoring as it has the appropriate sensitivity to detect a relapse at a very early stage.
Can I use INVIEW Oncopanel All-in-one Liquid Biopsy for diagnostic purposes?
The product is available for research use only (RUO). All samples are processed compliant to ISO 17025:2005 accreditation.
What are the limitations of the CNV analysis?
Even when control samples (plasma samples from at least seven other patients/individuals) are submitted, the CNV analysis approach could be subjected to limitations. The sensitivity and specificity of the assay will depend on the level of CNVs present and the tumour fraction.
Will all copy number variations in my sample be detected?
Tumour heterogeneity could make it difficult to correctly identify all relevant copy number variations in a given plasma sample. In addition, excessive contamination with DNA from normal cells could lead to coverage differences that are too small to be detected even with Eurofins Genomics highly sensitive methods.
Where do I get my results?
All deliverables (FAST-Q files and analyzed data) can be downloaded via your secure Ecom online account.
How soon after the blood draw should I ship the sample?
We recommend that the blood sample be shipped to Eurofins Genomics within 24 hours after blood drawing. The sample must be stored in BCT Streck tubes. Storage and delivery of blood samples must be at room temperature. Plasma samples have to be shipped on dry ice.
Where should I send my samples?
Eurofins Genomics Europe Pharma and Diagnostics Products & Services Sanger/PCR GmbH
Jakob-Stadler-Platz 7
78467 Konstanz
Germany
Questions on Inview Oncopanel All-In-One
What kind of samples can be processed with Inview Oncopanel All-In-One?
For the INVIEW Oncopanel, we accept fresh-frozen and FFPE tissue and genomic DNA.
For liquid biopsy analysis of cell-free DNA (cfDNA) isolated from blood plasma, please refer to our GATCLiquid Oncopanel All-In-One product.
How is the target enrichment performed?
The genes of interests are enriched via a proprietary Eurofins protocols. Target enrichment approach outcompetes common PCR-based enrichment with very uniform coverage and all exons of the genes covered. The proven target enrichment method has been optimised by Eurofins to develop highly complex libraries from low DNA and to deliver deep and uniform sequence coverage. Following target capture, next-generation sequencing of the library is performed.
What kind of mutations can be detected with Inview Oncopanel All-In-One?
The service fully covers the entire exons of about 600 cancer-relevant genomic regions, including protein-coding genes, select promoter regions, miRNAs, extra-exonic variants and select fusion gene events. In addition to SNPs and InDels, the product can also detect CNVs.
How is the tumor mutational burden (TMB) calculated?
- TMB is defined as the number of somatic, coding, base substitution, and InDel mutations per megabase of genome examined. All base substitutions and InDels in the coding region of targeted genes, including synonymous mutations, are initially counted before filtering as outlined below.
- The following mutations are excluded in silico from the TMB computation: Known somatic mutations in COSMIC and ClinVar, low-confident mutations, known germline variants in the ExAC (gnomAD) database, mutations predicted to be germline by the somatic-germline-zygosity algorithm, and mutations in tumor suppressor genes due to the focus of the Oncopanel All-In-One on actionable cancer mutations and potential panel design bias.
- To calculate the TMB per megabase, the total number of mutations counted is normalized by the size of the coding region of the targeted region in megabase (mutations per megabase, mut/MB). Due to the lack of standardization in TMB quantification we provide three TMB values:
- Non-synonymous mutations
- Non-synonymous mutations and plus indels and iii) including all mutations.
Is a focused gene panel, like Oncopanel All-In-One, suitable for TMB determination in comparison to whole-exome sequencing (WES)?
Although the gold standard for TMB determination is whole-exome sequencing (WES), focused panels, like the Oncopanel All-In-One v2.0, have been evaluated extensively for TMB estimation because of their lower sequencing cost and higher sensitivity. Recent research shows, that panels with a size > 1.5 Mbp are sufficiently suited to determine TMB with a high concordance to WES (Buchhalter et al. 2019, Int J Cancer 144:848–858). Thus, Oncopanel All-In-One 2.0’s size of 3 Mbp provides more precise TMB estimates than other smaller panels.
What is the sensitivity of the product?
SNP and InDel detection has a technical sensitivity down to 1%.
What kind of quality controls do you perform?
The quality and quantity of each sample will be determined at sample receipt or after DNA extraction. Further quality controls are performed at various steps of the process.
What is the difference between Inview Oncopanel All-In-One and GATCLiquid Oncopanel All-In-One?
Both tests offer cancer variant detection services based on a validated oncology panel. The interrogated genes and genomic alterations of the panel are the same. Similarly, the techniques used for both products are based on hybridisation for target enrichment and next-generation sequencing.
The two products differ in their starting material. Inview Oncopanel All-In-One can accept conventional source material like fresh-frozen and FFPE tissues, whereas GATCLiquid Oncopanel All-In-One analyses cfDNA extracted from blood plasma samples.
Can I directly compare results between Inview Oncopanel All-In-One and GATCLiquid Oncopanel All-In-One?
Yes, the two services are ideally suited for studies aimed at comparing the performance of traditional biopsies versus liquid biopsies.
What are the limitations of the CNV analysis?
Even when matched samples (tumour tissue and normal tissue or blood from the same patient) are submitted, the paired analysis approach could be subjected to limitations. The measurement variance on this single matched reference sample will be higher than that for a reference consisting of multiple reference samples. This could cause a modest number of (false-positive) CNV calls even in cases where the two paired samples are truly copy number identical.
Will all variants in my sample be detected?
Tumour heterogeneity could make it difficult to correctly identify all relevant copy number variations in a given sample. Even when the tumour itself is homogeneous, excessive contamination of normal cells within a tumour sample could lead to coverage differences that are too small to be detected even with Eurofins Genomics highly sensitive methods.
Can I use Inview Oncopanel All-In-One for diagnostic purposes?
The product is available for research use only (RUO). The service is performed in an ISO17025:2005 –accredited and ISO13485:2016-certified laboratory.
Where do I get my results and in which form are they delivered?
All deliverables (FAST-Q files and analyzed data) can be downloaded via your secure online account.
Where should I send my samples?
Eurofins Genomics Europe Pharma and Diagnostics Products & Services Sanger/PCR GmbH
Jakob-Stadler-Platz 7
78467 Konstanz
Germany
Questions on Inview Metagenome Advance
What kind of samples can be used?
For Inview Metagenome Advance we accept clinical research samples of human origin (e.g. swaps, feces, lavage). Upon request many types of starting material can be used for whole metagenome analysis such as:
food samples for quality control and pathogen detection in an industrial environment (dairies, breweries, meat-processing and agricultural facilities)
environmental samples
Eurofins Genomics is particularly well-experienced in DNA isolation and sequencing of stool samples.
How much starting material is required?
For specific information on quantity and concentration requirements for specific library preparation methods please refer to your quote or contact us directly. For best results we typically require >100 ng of double-stranded, purified DNA (total amount depends on the proportion of microbial DNA in the sample). Please note that the DNA has to be RNA-free with an OD 260/280 >1.8 and OD 260/230 >2.0.
What data will I receive?
Eurofins Genomics will provide you with tables (tsv, txt) that list all genera present in your sample and the respective read counts. In addition, you will receive all raw sequencing data.
Operational Taxonomic Units (OTUs), which are classified by Kraken’s exact k-mer alignment algorithm and appropriate MiniKrakenDB database, are represented in table format with taxonomic identification.
What is the necessary coverage for metagenome analysis?
The required sequencing depth for successful metagenome sequencing mainly depends on the complexity of the sample (number and representation of individual species) and the level of expected host contamination. Microbial communities of high complexity with high background levels of host DNA require more coverage than samples of reduced complexity and with lower host DNA contamination levels. In case of doubt we recommend performing a pilot on a sub-set of samples to determine the required sequencing coverage.
Do you guarantee a certain sequencing output?
Yes, Inview Metagenome Advance comes with guaranteed 10 million read-pairs. Additional read packages can be ordered separately.
Which organisms can be detected?
Metagenomics analysis permits the identification of microorganisms independent of taxonomic markers such as 16S rRNA. Metagenome sequencing enables the identification of both culturable and unculturable organisms, such as bacteria, archaea, fungi, protozoa and viruses. Although Eurofins Genomics is most experienced in profiling human microbiota, we have also successfully characterised microbial community members from food, industrial and environmental samples.
How do I choose between targeted amplicon sequencing and metagenome sequencing?
Both amplicon and metagenome sequencing present distinct advantages as well as disadvantages. Your method of choice should be based on your research goal. The successful outcome of a project typically depends on several factors such as community composition, the abundance of closely-related species and sequencing depth.
Amplicon sequencing offers suitable taxonomic profiling of a large number of samples. The approach enables the detection of subtle differences between microbial communities, which makes the method beneficial for statistical comparisons, such as for case control studies or for sampling varying environments over time.
Metagenomic sequencing does not rely on a certain set of primers, which frees the method of taxon-specific PCR biases. This makes possible more accurate representations of the analysed microbiota and more dependable estimations species abundance. Metagenomics analysis can provide information on both the taxonomic as well as the functional characteristics of a given sample.
What is the difference between Inview Metagenome Explore and Inview Metagenome Advance?
Inview Metagenome Explore is a ideally suited for broad taxonomic profiling of all organisms present in a given microbiome. The included 10 million read pairs are suitable for analysis of low complexity metagenomes, as well as for a broad overview of genera in complex metagenomes. A deeper sequencing coverage can be accomplished by adding additional read packages.
Inview Metagenome Advance provides detailed assessment of antibiotic resistance and community function, in addition to taxonomic characterisation. The product is ideal for profiling of complex metagenomes. For a deeper sequencing coverage than the included 10 million read pairs additional read packages can be added. The service can be applied to the quantification of functional processes in a particular community or to the detection of numerous resistance factors in an undisturbed natural environment. Please note that the required sequencing depth for successful metagenomic profiling is strongly influenced by the complexity and expected level of host contamination of the starting material. Therefore, additional sequencing effort could be necessary to obtain satisfactory results.
Where should I send my samples?
Eurofins Genomics Europe Pharma and Diagnostics Products & Services Sanger/PCR GmbH
Jakob-Stadler-Platz 7
78467 Konstanz
Germany
How should I send my samples?
- Please use 1.5 ml safe-lock tubes for your templates and primers
- Do not tape or wrap tubes with parafilm. Safe-lock tubes offer perfect sealing and evaporation protection
- Label your template with our free NGS barcode labels.
If you send more than 92 samples then you can also sent the samples in plates. Please use full skirted PCR plates (e.g. Eppendorf or Twin).
For more information and for sample submission for extraction please read our Sample Submission Guide.
(Picture shows the procedure with Sanger Prepaid Barcodes but work the same with free NGS Barcodes)
|
1
Only use 1.5 ml
safe-lock tubes
|
2
Remove barcode
stricker from film
|
3
Affix barcode sticker
horizontally
|
4
QR-code visible at front so
it can be read by scaner
|
Questions on Inview Metagenome Explore
What kind of samples can be used?
In general all kind of (DNA) samples can be used for metagenome profiling such as:
- samples of human origin (swaps, feces, lavage)
- food samples for quality control of microorganisms and pathogen detection in an industrial setting (dairies, breweries, meat-processing and agricultural facilities)
- environmental samples
Eurofins Genomics is particularly well-experienced in DNA isolation and sequencing of stool samples.
How much starting material is required?
For information on quantity and concentration required for specific library preparation methods please refer to your quote or contact us. For optimal results we generally require >100 ng of double-stranded, purified DNA (total amount depends on the proportion of microbial DNA in the sample). Please note that the DNA has to be RNA-free with an OD 260/280 >1.8 and OD 260/230 >2.0.
What data will I receive?
Eurofins Genomics will deliver tables (tsv, txt) listing all genera present in the analysed sample and the respective read counts. In addition, you receive all raw sequencing data.
Operational Taxonomic Units (OTUs), which are classified by BLAST analysis against appropriate databases, are represented in a tabular form including taxonomic identification. Only the best hits are considered for OTU assignment.
What is the necessary coverage for metagenome analysis?
The required sequencing depth for successful metagenome sequencing mainly depends on the complexity of the sample (number and representation of individual species) and the level of expected host contamination. Samples of high complexity with high background levels of host DNA require more coverage than samples of reduced complexity and with lower host DNA contamination levels. In case of doubt we recommend performing a pilot on a sub-set of samples to determine the required sequencing coverage.
Do you guarantee a certain sequencing output?
Yes, Inview Metagenome Explore comes with guaranteed 10 million read-pairs. Additional read packages can be ordered separately.
Which organisms can be detected?
Metagenomics analysis permits the identification of microorganisms independent of taxonomic genetic markers. The approach enables the identification of culturable and unculturable organisms, such as bacteria, archaea, viruses, as well as simple eukaryotes including fungi and protists. Although Eurofins Genomics has the most expertise in profiling human microbiota, we have also successfully identified microbial community members from food, industrial and environmental samples.
How do I choose between targeted amplicon sequencing and metagenome sequencing?
Both amplicon and metagenome sequencing have their own advantages and disadvantages.
Your method of choice should be selected based on your research aim. Generally, the successful outcome of a project depends on several factors such as community composition, the abundance of other close species or strains and sequencing coverage depth.
Amplicon sequencing offers suitable assessment of taxonomic composition and diversity of a large number of samples. As you have more power to detect subtle differences between microbial communities, the method is beneficial for statistical comparisons, such as for case control studies or for sampling environments over time.
Metagenomic sequencing is free of taxon-specific PCR biases due to the primer sets used. This enables more accurate representations of the analysed microbiota and more dependable estimations of species abundance levels. Metagenomics analysis can also help elucidate both taxonomic as well as functional characteristics of a given sample.
What is the difference between Inview Metagenome Explore and Inview Metagenome Advance?
Inview Metagenome Explore is a great product for the general taxonomic profiling of all organisms present in a given microbiome. The included 10 million read pairs are suitable for analysis of low complexity metagenomes, as well as for a broad overview of genera in complex metagenomes. A deeper sequencing coverage can be accomplished by adding additional read packages.
Inview Metagenome Advance offers detailed assessment of antibiotic resistance and community function, in addition to taxonomic characterisation. The product is ideal for profiling of complex metagenomes. For a deeper sequencing coverage than the included 10 million read pairs additional read packages can be added. The service can help quantify functional processes in a particular community or detect numerous resistance factors in an undisturbed natural environment.
Where should I send my samples?
Eurofins Genomics Europe Pharma and Diagnostics Products & Services Sanger/PCR GmbH
Jakob-Stadler-Platz 7
78467 Konstanz
Germany
Questions on INVIEW Transcriptome (Illumina)
Which INVIEW Transcriptome package do you recommend for which application?
This depends on many factors, such as tissue type, sample quality, development status, existing references, etc. Please refer to the table shown under Product Specifications to find your optimum INVIEW Transcriptome solution. The estimated read numbers shown there refer to human samples. Alternatively, please contact us to discuss your project. In some cases it might be advisable to set up a trial to define how many reads will be required for your specific aim or organism.
INVIEW Transcriptome Discover uses a strand-specific RNA library combined with Illumina’s 150 bp paired-end sequencing and is therefore recommended for the detection of differentially expressed genes, rare and novel transcripts and for discovering splice variants. In addition, the information about the transcript’s orientation allows for a more precise determination of structure and gene expression.
RNA-Seq of bacterial samples most often requires less reads since no splice variants are present. The INVIEW Transcriptome Bacteria is a cost-alternative for all projects interested in the gene expression profile of Bacteria.
The INVIEW Transcriptome Ultra Low is specially designed for all projects with only a tiny amount of starting material.
Do I need a genomic reference sequence?
INVIEW Transcriptome Discover:
Not necessarily. However, if you have a reference, please refer to the recommendations below.
INVIEW Transcriptome Bacteria / Ultra Low:
A clearly defined Ensembl name (e.g., GRCh37 or Rnor_5.0) for the annotated genomic reference sequence has to be provided prior to project start. Alternatively, the genome sequence can be provided in Fasta (along with the corresponding annotation in Gene transfer format (gtf)) or in GenBank format (including annotation).
Which starting material will be accepted?
Total RNA from tissues, cells or body fluids from eukaryotic or prokaryotic organisms can be used to prepare libraries for sequencing. In addition to RNA isolated from fresh tissue, RNA from FFPE samples can be analysed. RNA isolation can be offered as an additional service. For starting material from other sources please contact us.
INVIEW Transcriptome Ultra Low:
Please note that we cannot accept cells in cell culture media for the INVIEW Transcriptome Ultra Low package.
How much starting material is required?
For information on the quantity and concentration of RNA required for specific library preparation methods, please refer to your quote or contact us. In general, we require 150 ng of high-quality RNA (up to 25 µl / optimal concentration 6 - 50 ng/µl (absolute maximum concentration 200 ng/µl)) with an OD 260/280 of 1.8-2.0 and an OD 260/230 of 2.0-2.2 for optimum results. It might be possible to start with less material, but this depends a great deal on the quality of the material and the requested library.
The quality and quantity of the provided starting material are crucial for the success of sample preparation. The use of too little, degraded, contaminated or otherwise damaged starting material can result in a low yield or sample preparation failure and impair the quality and quantity of sequencing results.
INVIEW Transcriptome Ultra Low:
Minimum 0.15 ng/µl (in min. 10 µl); RIN > 6.5
Can RNA isolation be offered?
Eurofins Genomics offers RNA isolation as an additional service. We have successfully optimised protocols for extraction of high-quality RNA from several sample types and starting materials. Please refer to Product Specifications for more information on RNA isolation or consult us directly to discuss possible sample preparation methods for your individual requirements.
Which starting materials and aims are best suited for performing ribosomal RNA depletion?
a) For eukaryotes: rRNA depletion is recommended for partially degraded RNA or for when sequencing RNA with no poly-A tail.
b) For prokaryotes we recommend rRNA depletion in general, because poly-(A) enrichment of prokaryotic transcripts is not feasible.
You can order rRNA depletion from Eurofins Genomics. Or, alternatively, you can provide us with RNA that has already been rRNA depleted / mRNA enriched as starting material (20 ng per sample (up to 15 µl / concentration >1,4 ng/µl)). Please see Product Specifications for more information.
How efficient is ribosomal RNA depletion?
The efficiency of removal is determined by the organism, the composition and the quality of your sample RNA. If the RNA is severely degraded, ribodepletion might be less efficient.
Should I send my starting material on dry ice or would room temperature be sufficient?
RNA must be shipped on dry ice, unless the RNA is precipitated in ethanol. Tissues / cell cultures must be flash frozen in liquid nitrogen or dry ice and have to be shipped on dry ice. Alternatively, fresh material can be stabilised in “RNAlater” (e.g., Ambion, SIGMA or QIAGEN) and be sent at room temperature. Please verify in advance that the couriers you are using accept dry-ice shipments.
What should I do if my sample fails the entry QC?
If the amount, concentration or quality of the starting material does not meet requirements for further processing, we will contact you to discuss how to proceed further. If possible, Eurofins Genomics will recommend additional pre-processing steps in order to optimise sample quality.
Where and in what form can I obtain my results?
All raw data (FAST-Q files) as well as the analysed data can be downloaded via your secure online account.
What kind of BioIT do you offer?
Affordable high-quality bioinformatics analysis, including data filtering and QC, is optionally available for each package.
Where should I send my samples?
Eurofins Genomics Europe Pharma and Diagnostics Products & Services Sanger/PCR GmbH
Jakob-Stadler-Platz 7
78467 Konstanz
Germany
How should I send my samples?
- Please use 1.5 ml safe-lock tubes for your templates and primers
- Do not tape or wrap tubes with parafilm. Safe-lock tubes offer perfect sealing and evaporation protection
- Label your template with our free NGS barcode labels.
If you send more than 92 samples then you can also sent the samples in plates. Please use full skirted PCR plates (e.g. Eppendorf or Twin).
For more information and for sample submission for extraction please read our Sample Submission Guide.
(Picture shows the procedure with Sanger Prepaid Barcodes but work the same with free NGS Barcodes)
|
1
Only use 1.5 ml
safe-lock tubes
|
2
Remove barcode
stricker from film
|
3
Affix barcode sticker
horizontally
|
4
QR-code visible at front so
it can be read by scaner
|
Questions on NGSelect Amplicon
What starting material can be accepted?
For a list of accepted starting material, including the relevant amounts, please refer to Table 1 in Product Specifications.
What kind of quality control do you perform?
NGSelect Amplicon on NovaSeq (150 - 270 bp):
Prior to sequencing, the quality and quantity of each sample will be determined by established methods (e.g. agarose gel analysis / Qubit® Fluorometer / NanoDrop / Agilent 2100 Bioanalyzer). Further quality controls are performed at various steps of the sequencing workflow.
NGSelect Amplicon on MiSeq (280 - 570 bp):
Beside the evaluation of the gel picture you are sending, the success of the PCR reaction serves as our quality control. After the library prep and at various steps of the sequencing workflow further quality controls are performed.
What kind of BioIT do you offer?
Affordable high quality bioinformatics analysis is available for each package (see “Product Specifications” for more.
Can I use the results for diagnostic purposes?
Yes. On request, the service can be performed under diagnostic conditions with ISO17025 certified processes.
Where do I get my results and in which form are they delivered?
The results will be delivered via your Ecom account.
Where should I send my samples?
Eurofins Genomics Europe Pharma and Diagnostics Products & Services Sanger/PCR GmbH
Jakob-Stadler-Platz 7
78467 Konstanz
Germany
For the product NGSelect Amplicon on MiSeq, can we send samples that consist of several amplicons generated with different primer pairs?
Usually you would send per sample 1 amplicon generated with 1 primer pair. In some cases you might require however less reads per amplicon as given in the Project Specification. In such cases the individual amplicon samples you send may also consist of several amplicons. Please note, that in this case however we will not perform the clipping of primer sequences and deliver raw data only.
How should I send my samples?
- Please use 1.5 ml safe-lock tubes for your templates and primers
- Do not tape or wrap tubes with parafilm. Safe-lock tubes offer perfect sealing and evaporation protection
- Label your template with our free NGS barcode labels.
If you send more than 92 samples then you can also sent the samples in plates. Please use full skirted PCR plates (e.g. Eppendorf or Twin).
For more information and for sample submission for extraction please read our Sample Submission Guide.
(Picture shows the procedure with Sanger Prepaid Barcodes but work the same with free NGS Barcodes)
|
1
Only use 1.5 ml
safe-lock tubes
|
2
Remove barcode
stricker from film
|
3
Affix barcode sticker
horizontally
|
4
QR-code visible at front so
it can be read by scaner
|
Questions on NGSelect DNA
What starting material can be accepted?
For a list of accepted starting material, including the relevant amounts, please refer to Table 1 in Product Specifications.
What kind of quality control do you perform?
The quality and quantity of each incoming sample will be determined by appropriate methods (e.g. agarose gel analysis / Qubit® Fluorometer / NanoDrop / Agilent 2100 Bioanalyzer). Further quality controls are performed at various steps of the process.
What should I do if my sample fails the entry QC?
If the amount, concentration and / or quality of the starting material do not meet the requirements for further processing, we will contact you to discuss how to proceed. If possible, Eurofins Genomics will recommend additional pre-processing steps in order to optimise the sample quality.
What kind of BioIT do you offer?
Affordable high quality bioinformatics analysis is available for each package including data filtering and QC.
Can I use the results for diagnostic purposes?
Yes, upon request, the service can be carried out under diagnostic conditions with ISO17025 certified workflows.
Where do I get my results and in which form are they delivered?
All raw data (FAST-Q files) as well as the analysed data can be downloaded via your secure online account.
Where should I send my samples?
Eurofins Genomics Europe Pharma and Diagnostics Products & Services Sanger/PCR GmbH
Jakob-Stadler-Platz 7
78467 Konstanz
Germany
How should I send my samples?
- Please use 1.5 ml safe-lock tubes for your templates and primers
- Do not tape or wrap tubes with parafilm. Safe-lock tubes offer perfect sealing and evaporation protection
- Label your template with our free NGS barcode labels.
If you send more than 92 samples then you can also sent the samples in plates. Please use full skirted PCR plates (e.g. Eppendorf or Twin).
For more information and for sample submission for extraction please read our Sample Submission Guide.
(Picture shows the procedure with Sanger Prepaid Barcodes but work the same with free NGS Barcodes)
|
1
Only use 1.5 ml
safe-lock tubes
|
2
Remove barcode
stricker from film
|
3
Affix barcode sticker
horizontally
|
4
QR-code visible at front so
it can be read by scaner
|
Questions on NGSelect Ready-to-load
What starting material can be accepted?
For a list of accepted starting material, including the relevant amounts, please refer to Table 1 in Product Specifications.
What kind of quality control do you perform?
All ready-to-load libraries received are subject to a quality and quantity control to accurately determine the appropriate sequencing dilution and achieve optimal cluster densities.
What should I do if my sample fails the entry QC?
If the amount, concentration or quality of the ready-to-sequence library does not meet the requirements for further sample processing, we will contact you to discuss how to go on with the project. Whenever possible, Eurofins Genomics will make recommendations for optimisation of sample quality.
What kind of BioIT do you offer?
Affordable and professional bioinformatics analysis is available for on request. For more information, see “Product specifications”.
Can I use the results for diagnostic purposes?
Yes, on request, the sequencing service can be performed under diagnostic conditions that are ISO17025 accredited.
Where do I get my results and in which form are they delivered?
All raw data (FAST-Q files) as well as the analysed data can be downloaded via your secure online account.
Do you guarantee a certain number of reads?
The actual achieved sequencing output (read length, read quality and number of reads) is directly correlated with the quality of the sequencing library used. Eurofins Genomics cannot give any guarantees for sequencing data resulting from ready-to-load libraries prepared by the customer. If unsatisfactory sequencing results are due to technical problems with the sequencing kits or machines, the sequencing will be repeated at no additional cost to the customer.
Is the sorting of c included?
Raw data sorting according to Illumina Index read is included. Used index type (single-indexed or dual-indexed) and corresponding index sequences have to be provided prior to project start.
Raw data sorting according to customer specific tags at read start can be offered at additional costs. We highly recommend to avoid the “in-line” barcoding strategy and use the Illumina index system instead (i.e. the barcodes are read in a separate read and do not interfere with cluster registration). It is important to ensure that the base composition of the indices is balanced to optimise the ability of the image analysis software to distinguish signals.
What steps are taken to increase sequencing quality?
Illumina cluster detection algorithms are optimized around a balanced representation of A, T, G, and C nucleotides. Any divergence from equal base distribution will negatively influence the amount and quality of sequencing data produced. To increase the library nucleotide balance a spike-in of 20% PhiX will be used for samples with low diversity or unbalanced base composition (e.g., amplicons, bisulfite converted samples). The extent to which the negative impact of the unbalanced base composition will be reduced by spiking the PhiX control depends on individual sample and sequence characteristics. For more information please refer to the Illumina website.
How can I choose between the different NGSelect services?
NGSelect is divided into four major categories, depending on starting material, DNA, RNA, amplicons or ready-to-load libraries. Each service offers a wide range of options that help create individualised and affordable NGS projects. Please see table 2 (below) for more information:

Questions on INVIEW Oncoexome Liquid Biopsy
What kind of samples can be processed with INVIEW Oncoexome Liquid Biopsy?
For INVIEW Oncoexome Liquid Biopsy, we accept fresh or retained plasma samples as well as already isolated cell-free DNA. Please refer to our Liquid Biopsy sample preparation sheet (see "Files"). The ctDNA is extracted from the blood plasma fraction.
For starting material such as DNA isolated from FFPE and tissue samples, please refer to our product Inview Human Exome.
How much starting material is required?
In general we require either 4 mL of plasma or 15 ng already isolated cell-free DNA (up to 30 µl / concentration > 0,5 ng/µl).
What pre-sequencing services are available?
Several pre-sequencing services are included in the Oncoexome product. These include ctDNA extraction from plasma and high- complexity library preparation with low duplicate rates. Exon targeting is accomplished with Agilent’s SureSelect platform, which offers superior exon enrichment and coverage uniformity.
What sequencing coverage should I use?
The required coverage depends on the required sensitivity of variant detection. You may choose the sequencing coverage you need for your individual project study objective.
We typically recommend 100x coverage based on experience gained from analysing cell-free DNA from more than 40,000 samples to date.
What kind of quality controls do you perform?
The quality and quantity of each sample will be determined upon receiving the sample. Further quality controls are performed at various steps of the process.
How should I choose between Oncoexome, Oncopanel and Oncotarget?
All three tests are based on liquid biopsy. The three products serve as powerful non-invasive tools for real-time cancer profiling with the ability to capture the entire heterogeneity of the disease.
INVIEW Oncoexome Liquid Biopsy is the first commercially available service for whole exome sequencing (WES) of ctDNA. With this service, you can obtain a comprehensive profile of all mutations present in the protein-coding regions of the genome. This product is suitable for a preliminary look at the genome of a suspected cancer patient, in cases where limited information regarding clinically relevant mutations is available.
INVIEW Oncopanel Liquid Biopsy is a comprehensive NGS panel for profiling important cancer mutations. It uses single-molecule PCR to target 50 known cancer genes including tumour suppressors, mutation hotspots and drug-resistance markers. The panel sets a new industry standard in terms of sequence coverage and uniformity with a sensitivity of about 1%.
INVIEW Oncotarget facilitates ultra-sensitive detection of clinically relevant cancer-specific point mutations, insertions, deletions and gene fusions in circulating tumour DNA down to an allele frequency of 0.1%. This approach is suitable for treatment monitoring as it is sensitive enough to detect a relapse at a very early stage.
How do I obtain my results and what form will they be in when delivered?
All deliverables (FAST-Q files and analyzed data) can be downloaded via your secure online account.
How soon after the blood draw should I ship the sample?
Blood samples have to be shipped to Eurofins Genomics within 24 hours after drawing blood. The sample must be stored in BCT Streck tubes. Samples may be stored and delivered at room temperature. Plasma samples have to be shipped on dry ice.
How many samples can I submit?
Eurofins Genomics accepts any number of samples in multiples of four.
Where should I send my samples?
Eurofins Genomics Europe Pharma and Diagnostics Products & Services Sanger/PCR GmbH
Jakob-Stadler-Platz 7
78467 Konstanz
Germany
Questions on NGSelect RNA
What starting material can be accepted?
For a list of accepted starting material, including the relevant amounts, please refer to Table 1 in Product Specifications.
Do I always need a genomic reference sequence?
Typically, a clearly defined Ensembl name for the annotated genomic reference sequence has to be provided before the project starts. Another option is to provide the genome sequence in Fasta format (along with the respective annotation in Gene transfer format (gtf)) or in GenBank format (including annotation).
What kind of quality control do you perform?
The quality and quantity of each sample is determined by appropriate methods (e.g. agarose gel analysis / Qubit® Fluorometer / NanoDrop / Agilent 2100 Bioanalyzer). Further quality controls are performed at nearly all steps of the process.
What should I do if my sample fails the entry QC?
If the amount, concentration or quality of the starting material does not meet the requirements for further sample processing, we will contact you to discuss how to go on with the project. If possible, Eurofins Genomics will make recommendation son how to optimise sample quality.
What kind of BioIT do you offer?
Affordable high quality bioinformatics analysis is available for each package including data filtering and QC.
Can I use the results for diagnostic purposes?
Yes, upon request, the service can be carried out under diagnostic conditions with ISO17025 certified workflows.
Where do I get my results and in which form are they delivered?
All raw data (FAST-Q files) as well as the analysed data can be downloaded via your secure online account.
Where should I send my samples?
Eurofins Genomics Europe Pharma and Diagnostics Products & Services Sanger/PCR GmbH
Jakob-Stadler-Platz 7
78467 Konstanz
Germany
How should I send my samples?
- Please use 1.5 ml safe-lock tubes for your templates and primers
- Do not tape or wrap tubes with parafilm. Safe-lock tubes offer perfect sealing and evaporation protection
- Label your template with our free NGS barcode labels.
If you send more than 92 samples then you can also sent the samples in plates. Please use full skirted PCR plates (e.g. Eppendorf or Twin).
For more information and for sample submission for extraction please read our Sample Submission Guide.
(Picture shows the procedure with Sanger Prepaid Barcodes but work the same with free NGS Barcodes)
|
1
Only use 1.5 ml
safe-lock tubes
|
2
Remove barcode
stricker from film
|
3
Affix barcode sticker
horizontally
|
4
QR-code visible at front so
it can be read by scaner
|
Questions on INVIEW Microbiome Profiling 3.0
What Species can be detected using Microbiome Profiling by NGS?
Depending on your choice of target regions, all bacterial, archeal and fungal species of which a sequence is published in the NCBI nucleotide database can be identified. There are currently for example thousands of bacterial species with sequence entries in the database. Closely related species with almost identical DNA sequences will not be discriminated and only the genus (or the family) will be reported.
What are the limitations of using Microbiome Profiling by NGS?
• Closely related species are not distinguishable because of sequence identity.
• Degradation of bacterial DNA may occur in highly processed samples or samples with low pH - a species identification is therefore difficult and sometimes not possible.
• The more starting material we receive, the higher the probability to get excellent sequencing results.
What is the level of false positives / negatives?
• Samples with strongly degraded DNA will give the result “no species identification possible”. To avoid false-positive results, a negative control (water) is always co-analysed with the samples.
• With NGS all DNA amplicons that were successfully amplified from the sample DNA will be detected. Due to the high sensitivity of the method, sequences with low presence in the sequence pool or sequences with unexpected sequence lengths have to be removed from the data analysis during the bioinformatical workflow in order to exclude false-positives due to PCR and/or sequencing errors. Species with a fraction below 0.5 % of all assigned reads are not reported.
How should I choose the target regions and the amount of targets to be analysed?
For bacterial profiling please choose at least on 16S target, for fungal profiling at least one ITS target. For taxonomic profiling or archea please selcet our archeal 16S target.
As each primer set fails to amplify certain species we recommend that you carefully check the literature available about the limitations of each primer set. It is recommended to use multiple primer combinations to capture the entire community (e.g. Ihrmark et al., 2012; Toju et al., 2012).
What quality control do you apply on my samples?
We use the amplicon generation process as the entry quality control. Even though the PCR conditions are optimized the success of the amplicon generation depends on the amount of bacterial, archaeal or fungal DNA in the sample. In case we can not generate an amplicon or only a very week amplicon this is a strong indication that the content was too low. By default we will repeat the amplicon generation once with modified conditions.
What happens if no or only a weak amplicon can be generated from some of my samples:
In case no amplicon can be generated, we will stop processing of those samples and reimburse you for the sequencing part that was not performed. In the event that only a weak amplicon can be generated, Eurofins Genomics will include those amplicons in the amplicon pool for sequencing, too. Experience shows that those amplicons will result in very few reads only and interpretation of those reads should be done with special care.
When choosing your service on MiSeq, can we send samples that consist of several amplicons generated with different primer pairs?
Usually you would send per sample 1 amplicon generated with 1 primer pair. In some cases you might require however less reads per amplicon as given in the Project Specification. In such cases the individual amplicon samples you send may also consist of several amplicons. Please note, that in this case however we will not perform the clipping of primer sequences and deliver raw data only.
How should I send my samples?
- Please use 1.5 ml safe-lock tubes for your templates and primers
- Do not tape or wrap tubes with parafilm. Safe-lock tubes offer perfect sealing and evaporation protection
- Label your template with our free NGS barcode labels.
If you send more than 92 samples then you can also sent the samples in plates. Please use full skirted PCR plates (e.g. Eppendorf or Twin).
For more information and for sample submission for extraction please read our Sample Submission Guide.
(Picture shows the procedure with Sanger Prepaid Barcodes but work the same with free NGS Barcodes)
|
1
Only use 1.5 ml
safe-lock tubes
|
2
Remove barcode
sticker from film
|
3
Affix barcode sticker
horizontally
|
4
QR-code visible at front so
it can be read by scanner
|
Questions on INVIEW CRISPR Check
What kind of samples can be processed with INVIEW CRISPR Check?
The starting material is amplicons prepared in your lab that span the mutation site. The amplicon generation protocol to apply depends on the product type (adaptor ligation or 2nd PCR workflow), instructions can be found in the tab "How to prepare your Amplicons" on the respective product webpage. Amplicon length requirements can be found in the tab "Specifications".
Why is the recommended amplicon size range rather narrow?
For INVIEW CRISPR Check, the wildtype amplicon size needs to be adapted to the length of InDels you want to be able to analyse and must allow merging of read pairs. Usually mutations introduced via the NHEJ pathway are in a range of 1-20 bp, but in order to not miss the larger ones, the wildtype amplicons must be in a rather narrow size range. Length requirements for two use cases and all product types are available in our sample preparation guide.
Can I use your service if we used another CRISPR system than SpCas9?
Yes you can. In that case please provide the cleavage offset of your system (number of nucleotides upstream of PAM sequence where double strand break is expected).
What do you recommend for the assay setup?
- Always include at least one unmodified (wildtype) sample as control
- Include replicates if you aim for a very accurate prediction of the editing efficiency. Replicates increase accuracy of predicted editing efficiency, but are not strictly necessary.
How should I choose between INVIEW CRISPR Check Adaptor Ligation and INVIEW CRISPR Check 2nd PCR?
Both product types are excellent solutions to analyse mutations introduced by CRISPR/Cas system (NHEJ pathway).
The products vary mainly in the sequencing library preparation (2-step protocol vs. adaptor ligation protocol) and thus in the way you are preparing amplicons for sequencing.
A selection guide is available at the CRISPR website (tab Overview) to help you find the best solutions for your individual preferences.
What deliverables does your analysis provide?
- Comprehensive report with results and methods sections
- Raw FASTQ files
- Quality clipped and assembled FASTQ files
- Mapped reads in BAM format
- Observed wildtype and alternate allele sequences including their quantification
- Raw data, preprocessing, mapping, and coverage metrics in CSV format
- Calculated Editing Efficiency per sample
Where do I get my results?
For INVIEW CRISPR Check 2nd PCR the result files can be downloaded from a secure FTP server (password will be provided), for INVIEW CRISPR Check Adaptor Ligation the results will be delivered via your Ecom account.
Where should I send my samples?
Eurofins Genomics Europe Pharma and Diagnostics Products & Services Sanger/PCR GmbH
Jakob-Stadler-Platz 7
78467 Konstanz
Germany
When choosing INVIEW CRISPR Check 2nd PCR, can we send samples that consist of several amplicons generated with different primer pairs?Usually you would send per sample 1 amplicon generated with 1 primer pair.
In some cases you might require however less reads per amplicon as given in the Project Specification. In such cases the individual amplicon samples you send may also consist of several amplicons.
Please note, that in this case however we will not perform the clipping of primer sequences and deliver raw data only.
How should I send my samples?
- Please use 1.5 ml safe-lock tubes for your templates and primers
- Do not tape or wrap tubes with parafilm. Safe-lock tubes offer perfect sealing and evaporation protection
- Label your template with our free NGS barcode labels.
If you send more than 92 samples then you can also sent the samples in plates. Please use full skirted PCR plates (e.g. Eppendorf or Twin).
For more information and for sample submission for extraction please read our Sample Submission Guide.
(Picture shows the procedure with Sanger Prepaid Barcodes but work the same with free NGS Barcodes)
|
1
Only use 1.5 ml
safe-lock tubes
|
2
Remove barcode
stricker from film
|
3
Affix barcode sticker
horizontally
|
4
QR-code visible at front so
it can be read by scaner
|
Questions on INVIEW Virus
Do I always need a genomic reference sequence?
In case you order your NGS project with bioinformatic analysis (in this case: genome variant detection), then a reference genome is required.
Typically, a clearly defined Ensembl name for the annotated genomic reference sequence has to be provided before the project starts. Another option is to provide the genome sequence in Fasta format (along with the respective annotation in Gene transfer format (gtf)) or in GenBank format (including annotation).
What kind of quality control do you perform?
The quality and quantity of each sample is determined by appropriate methods (e.g. agarose gel analysis / Qubit® Fluorometer / NanoDrop / Agilent 2100 Bioanalyzer). Further quality controls are performed at nearly all steps of the process.
What should I do if my sample fails the entry QC?
If the amount, concentration or quality of the starting material does not meet the requirements for further sample processing, we will contact you to discuss how to go on with the project. If possible, Eurofins Genomics will make recommendation son how to optimise sample quality.
Can I use the results for diagnostic purposes?
Yes, upon request, the service can be carried out under diagnostic conditions with ISO17025 certified workflows.
Where do I get my results and in which form are they delivered?
All raw data (FAST-Q files) as well as the analysed data can be downloaded via your secure online account.
Where should I send my samples?
Eurofins Genomics Europe Pharma and Diagnostics Products & Services Sanger/PCR GmbH
Jakob-Stadler-Platz 7
78467 Konstanz
Germany
How should I send my samples?
- Please use 1.5 ml safe-lock tubes for your templates and primers
- Do not tape or wrap tubes with parafilm. Safe-lock tubes offer perfect sealing and evaporation protection
- Label your template with our free NGS barcode labels.
If you send more than 92 samples then you can also sent the samples in plates. Please use full skirted PCR plates (e.g. Eppendorf or Twin).
For more information and for sample submission for extraction please read our Sample Submission Guide.
(Picture shows the procedure with Sanger Prepaid Barcodes but work the same with free NGS Barcodes)
|
1
Only use 1.5 ml
safe-lock tubes
|
2
Remove barcode
stricker from film
|
3
Affix barcode sticker
horizontally
|
4
QR-code visible at front so
it can be read by scaner
|
Questions on INVIEW Plasmid Verification
What starting material can be accepted?
For a list of accepted starting material, including the relevant amounts, please refer to Table 1 in Product Specifications.
Where do I get my results and in which form are they delivered?
All raw data (FAST-Q files) as well as the analysed data can be downloaded via your secure online account.
Where should I send my samples?
Eurofins Genomics Europe Pharma and Diagnostics Products & Services Sanger/PCR GmbH
Jakob-Stadler-Platz 7
78467 Konstanz
Germany
In which format do I have to send the reference sequence?
The reference sequence can be uploaded, when accepting the quote in the sample questionnaire. Please make sure that the file does not exceed 2MB.
How should I send my samples?
- Please use 1.5 ml safe-lock tubes for your templates and primers
- Do not tape or wrap tubes with parafilm. Safe-lock tubes offer perfect sealing and evaporation protection
- Label your template with our free NGS barcode labels.
If you send more than 92 samples then you can also sent the samples in plates. Please use full skirted PCR plates (e.g. Eppendorf or Twin).
For more information and for sample submission for extraction please read our Sample Submission Guide.
(Picture shows the procedure with Sanger Prepaid Barcodes but work the same with free NGS Barcodes)
|
1
Only use 1.5 ml
safe-lock tubes
|
2
Remove barcode
stricker from film
|
3
Affix barcode sticker
horizontally
|
4
QR-code visible at front so
it can be read by scaner
|
Questions on Oxford Nanopore Sequencing
Which kind of services does Eurofins offer on the Oxford Nanopore instruments?
- Whole Genome Sequencing (WGS) of small genome
- Full-length 16S sequencing
- Amplicon sequencing
- Whole plasmid sequencing
Can I send already fragmented DNA?
No this is not recommended. Optimal results can be achieved when the fragmentation is done directly before the library preparation. We recommend to send unfragmented HMW DNA, as large as possible.
How should I send my samples?
- Please use 1.5 ml safe-lock tubes for your templates and primers
- Do not tape or wrap tubes with parafilm. Safe-lock tubes offer perfect sealing and evaporation protection
- Label your template with our free NGS barcode labels.
If you send more than 92 samples then you can also sent the samples in plates. Please use full skirted PCR plates (e.g. Eppendorf or Twin).
For more information and for sample submission for extraction please read our Sample Submission Guide.
(Picture shows the procedure with Sanger Prepaid Barcodes but work the same with free NGS Barcodes)
|
1
Only use 1.5 ml
safe-lock tubes
|
2
Remove barcode
stricker from film
|
3
Affix barcode sticker
horizontally
|
4
QR-code visible at front so
it can be read by scaner
|
Where should I send my samples?
Eurofins Genomics Europe Pharma and Diagnostics Products & Services Sanger/PCR GmbH
Jakob-Stadler-Platz 7
78467 Konstanz
Germany
Questions on OLINK
How many samples do I need to send?
The panels are handled in batches of 88 or 384 samples.
Where should I send my samples?
Eurofins Genomics Europe Pharma and Diagnostics Products & Services Sanger/PCR GmbH
Jakob-Stadler-Platz 7
78467 Konstanz
Germany
What sample types can be used with Olink Explore?
Olink Explore is fully validated for human plasma and serum samples.
Other sample types, for example CSF, have been shown to be compatible with the technology
For most other sample types, Olink® Target 96 panels are recommended, due to the longterm experience of the OLINK team with the target panel. Example sample types: tissue and cell lysates, fine needle biopsies, exosomes, microdialysis fluid, bronchoalveolar lavage fluid, cell culture media, dried blood spots, synovial fluid, cerebrospinal fluid, plaque extract, urine, saliva and many more.
How does the Olink® technology work?
The Olink® platforms are underpinned by a distinctive technology known as Proximity Extension Assay (PEA). This innovative dual-recognition, DNA-coupled methodology offers an unparalleled level of readout specificity. PEA empowers us to conduct high-throughput biomarker analysis with an extensive multiplex capability while maintaining uncompromising data quality standards.
Questions on bacterial genome sequencing
What is the turnaround time for bacterial genome sequencing?
On average, our turnaround time for bacterial genome sequencing, for genomes up to 7 Mb, is 3 to 5 business days.
Can I send my samples without the free barcode?
Unfortunately not. This barcode is required for our internal lab processes and is essential in ensuring that we can process your samples as efficiently as possible. Thank you for your cooperation!
Can I use my local dropbox to ship samples for Bacterial Genome Sequencing?
Yes of course.
If you don't know if there is a dropbox close by, use our dropbox location page to find out. Please click here >>
Troubleshooting guide for results
There are several reasons why your results might be different than what you expect.
To interpret your results you can have a look at our troubleshooting guide. Click here >>
Will you repeat sequencing of my sample if the sequencing fails?
For each run, we conduct multiple quality control (QC) measurements to assess the success of the library preparation and sequencing. If all the measurements indicate satisfactory results, we will not reprocess samples that have failed.
What level of accuracy can be expected from the results of bacterial genome sequencing?
According to the specifications provided by Oxford Nanopore for the v14 library prep chemistry and the R10.4.1 flowcells currently employed in bacterial genome sequencing, the raw read accuracy exceeds 99%. The final assembly's accuracy fluctuates based on coverage and data quality. Generally, higher coverage, implying more reads for consensus construction, tends to enhance result accuracy.
What are the expected outcomes or deliverables for a successful bacterial genome sequencing?
Successful sequencing is determined by attaining at least one of the following outcomes:
1. A well-constructed genome assembly.
2. The specified quantity of raw data (equivalent to about 30x genome coverage of an average bacterial genome).
Deliverables:
- Data analysis report in HTML format
- Raw Sequencing data
- Assembled genome in FASTA format
- Circular genome plot
- Annotated genome sequence of all contigs in GENBANK format.
- Annotated genome features in GFF3 format
- Annotated feature table, including the nucleotide sequence of each feature
- Nucleotide sequences of all gene sequences in FASTA format
- Amino acid sequences of all annotated protein sequences in FASTA format
Is it possible to sequence a mixture of bacterial species in one sample?
This service is designed for a clonal population (single strains) of bacteria. While you can submit mixtures of different bacterial strains for sequencing, it comes with a level of uncertainty in the assembly outcome, and therefore, it is done at your own risk.
If you are interested in mixed samples, we recommend using any of our other services:
Is it possible to sequence the native plasmids of my bacteria along with their genomic DNA (gDNA)?
If your genomic DNA (gDNA) extract includes native plasmid DNA, it is likely that you will receive sequencing reads for those plasmids. Generally, DNA fragments with a size of <1 kb are excluded during sequencing. However, small plasmid-sized reads are not filtered out during assembly, so there is a probability that they will contribute to their own plasmid assemblies in addition to the gDNA assembly.
Ultimately, the types of DNA in your sample that contribute to an assembly will depend on the overall sample quality, coverage, and the relative abundance or degradation of each type of DNA.
Can you also sequence genomes larger than 7 Mb or genomes that are linear or multichromosomal?
What is a contig?
A contig is the result of the assembly process, consisting of contiguous DNA sequences/ sequencing reads and not interrupted by unknown regions.
When my genome assembly looks perfect, can I assume that everything is correct?
Even though it appears 100% perfect, there is still a likelihood of having single nucleotide polymorphisms (SNPs) in your sequence, especially in regions with homopolymers where Oxford Nanopore technology tends to have a higher error rate. To achieve a truly flawless assembly, we recommend using additional Illumina data to polish your results. Please feel free to contact us for further assistance.
How about addressing the errors or positions with low confidence that I discovered in a homopolymer region or at a Dam or Dcm methylation site?
The prevalent error modes in Oxford Nanopore sequencing include deletions within homopolymer stretches, errors occurring at the central position of the Dcm methylation site CCTGG or CCAGG, and errors at the Dam methylation site GATC.
So please handle these regions with special care.
Questions on Clonal Amplicon Sequencing
Can I send my samples without the free barcode?
Unfortunately not. This barcode is required for our internal lab processes and is essential in ensuring that we can process your samples as efficiently as possible. Thank you for your cooperation!
Can I use my local dropbox to ship samples for Clonal Amplicon Sequencing?
Yes of course.
If you don't know if there is a dropbox close by, use our dropbox location page to find out. Please click here >>
Troubleshooting guide for results
There are several reasons why your results might be different than what you expect.
To interpret your results you can have a look at our troubleshooting guide. Click here >>
Will you repeat sequencing of my sample if the sequencing fails?
For each run, we conduct multiple quality control (QC) measurements to assess the success of the library preparation and sequencing. If all the measurements indicate satisfactory results, we will not reprocess samples that have failed.
How about addressing the errors or positions with low confidence that I discovered in a homopolymer region or at a Dam or Dcm methylation site?
The prevalent error modes in Oxford Nanopore sequencing include deletions within homopolymer stretches, errors occurring at the central position of the Dcm methylation site CCTGG or CCAGG, and errors at the Dam methylation site GATC.
So please handle these regions with special care.
Why are a few terminal nucleotides missing from my linear amplicon consensus sequence?
The assembler may encounter challenges reconstructing the terminal ends of linear DNA, leading to the potential omission of approximately ~10 nucleotides from the 3' and/or 5' ends of your insert, depending on your sample's sequence.
If you observe this issue with your samples, you can address it by downloading the raw reads from your Dashboard and reconstructing the ends using your preferred method.
What level of sequencing coverage can I expect for my clonal amplicons?
We do not provide a specific coverage guarantee. The number of raw reads generated can significantly vary based on sample quality. Typically, successful samples sent at the required concentration yield a range from high dozens to hundreds (or even thousands) of raw sequencing reads.
The average coverage of your sample is included in the result report HTML, where a coverage exceeding ~20x indicates a very accurate consensus.
Can I send mixtures of amplicons?
Our amplicon service is designed for a population of molecules that is clonal.
What will happen if you send mixtures?
If your species are sufficiently distinct (e.g. vastly different in size or sequence), the pipeline will first attempt to make a .fasta consensus file from the highest abundance species. It will also attempt to make a consensus of other species with read counts that are >20% of that of the most abundant species. Ultimately, which species end up producing a consensus will vary depending on overall sample quality, coverage, and relative abundance/degradation of each species.
How can I sequence my amplicon mixture?
Eurofins Genomics offers a vast amplicon sequencing portfolio. You will definitely find the correct service for your needs: Amplicon portfolio>>