Overview - What is whole-genome bisulfite sequencing (WGBS)?
One of the most commonly methylated sites in human DNA is 5-methylcytosine (5mC). In plants, 5mC is concentrated at CpG, CHG and CHH sequences (where H is either A, C or T). In vertebrates, 5mC is predominantly found at CpG islands, where the methylation functions in various processes such as gene expression regulation, embryonic development and carcinogenesis. Recent noninvasive diagnostic techniques are using the epigenetic state of cell-free DNA as a diagnostic biomarker for cancer.
Whole-genome bisulfite sequencing (WGBS) or bisulfite sequencing (Bisulfite-Seq or BS-Seq) combines bisulfite treatment with next-generation sequencing (NGS) for epigenetic analysis of the methylome of nearly any organism with single-base resolution. For vertebrates, this is the only technique that offers complete genome-wide coverage of CpGs. The method is considered the gold standard for epigenome profiling by the International Human Epigenome Consortium.
Applications - What are the advantages of BS-Seq?
Whole-genome bisulfite sequencing is considered the ultimate method for methylcytosine analysis, as the technique allows researchers to:
- Simultaneously observe the methylation patterns of all CpG, CHG and CHH sites present in the sample of interest
- Compare differentially methylated loci from different samples (ex.: healthy controls versus patients with cancer)
- Derive correlations between cytosine methylation and transcriptional regulation
- Investigate inherited methylations in nearly any organism
- Profile DNA methylations in the entire methylome for studies on development, cell differentiation, cell cycle, DNA repair, genomic imprinting
- Analyse the collective effect of methylation levels on cancer, autoimmunity, etc.
Workflow - Methods & technologies for WGBS
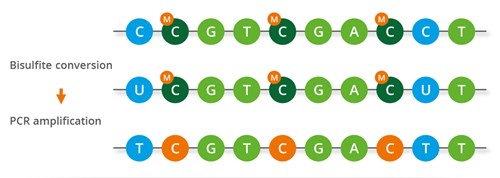
BS-Seq is the method of choice for obtaining a comprehensive analysis of the methylation patterns in the genome. The multi-step process employs bisulfite conversion, PCR amplification and next-generation sequencing to determine the methylation sites across the methylome.
Chemical treatment of DNA with sodium bisulfite converts unmethylated cytosines into uracils. Methylated cytosines are protected from bisulfite conversion by their methyl group, hence they remain unchanged during the conversion process. The DNA is then subjected to a PCR assay, where all uracils are converted to thymidines. The resulting DNA amplicons are sequenced with high sequencing coverage. The obtained sequencing reads are mapped against a reference genome to enable the identification of the methylated loci in the sample.
Methylation array vs. whole-genome bisulfite sequencing
Methylation arrays are a traditional technique for assessing the methylation status of cytosines. This method is used to target hundreds of thousands of methylation sites, rather than to probe the entire epigenome.
Focusing only on known methylated regions decreases the computational analysis of the assay; methylation arrays also suffer from significant setbacks compared to WGBS. Some of these disadvantages include:
- Limited possibilities for exploring organisms other than humans
- Inability to simultaneously detect both genetic mutations and epigenetic modifications
- Analysis of pre-determined genomic regions only
- Danger of cross-reactivity with non-targeted genomic regions or loci containing single nucleotide polymorphisms (SNPs)
- Extra computational analysis required for control of batch effects
Scientific expertise: BS-seq
Eurofins Genomics takes advantage of industry-leading platforms to offer extensive methylation analysis across the entire genome. Combined with thorough BioIT analysis, Eurofins Genomics can provide unmatched flexibility in delivering reliable data for achieving various research aims ranging from cancer research to understanding basic cellular processes.